A sufficient time had passed since my previous report such that there were both a number of things to report, and I crossed the required threshold of edginess to provide an update on the progress of the long list of things I am supposed to be doing in my quest to become Dr. Nicholls. I began a draft of this post shortly before I attended the European Conference on Computational Biology at the start of September. However at the end of the conference, I spontaneously decided to temporarily abort my responsibilities to bioinformatics, not return to Aberystwyth, electing to spend a few weeks traversing Eastern Europe instead. I imagine there will be more on this in a future post, but for now let’s focus on the long overdue insight into the work that I am supposed to be doing.
In our last installment, I finally managed to describe in words how the metahaplome is somewhat like a graph, but possesses properties that also make it not like a graph. The main outstanding issues were related to reweighting evidence in the metahaplome structure, the ongoing saga of generating sufficient data sets for evaluation and writing this all up so people believe us when we say it is possible. Of the many elephants in the room, the largest was my work still being described as the metahaplome or some flavour of Sam’s algorithm. It was time to try and conquer one of the more difficult problems in computer science: naming things.
Introducing Hansel and Gretel
Harbouring a dislike for the apparent convention in bioinformatics that software names should be unimaginative1 or awkwardly constructed acronyms, and spotting an opportunity to acquire a whimsical theme, I decided to follow in the footsteps of Goldilocks, and continue the fairy tale naming scheme; on the condition I could find a name that fit comfortably.
Unsurprisingly, this proved quite difficult. After some debate, the most suitable fairy tale name I could find was Rumpelstiltskin, for its ‘straw-to-gold’ reference. I liked the idea of your straw-like reads being converted to golden haplotypes. However as I am not remotely capable of spelling the name without Google, and the link between the name and software feels a tad tenuous, I vetoed the option and forged forward with the paper, with the algorithm Untitled.
As I considered the logistics of packaging the implementation as it stood, I realised that I had essentially created both a novel data structure for storing the metahaplome, as well as an actual algorithm for utilising that information to recover haplotypes from a metagenome. At this point everything fell into place; a nice packaging solution and a fitting pair of names had resolved themselves. Thus, I introduce the Hansel data structure, and the Gretel algorithm; a framework for recovering real haplotypes from metagenomes.
Hansel
Hansel is a Python package that houses our novel data structure. I describe it as a “graph-inspired data structure for determining likely chains of symbol sequences from crummy evidence”. Hansel is designed to work with counts of pairwise co-occurrences of symbols in space or time. For our purposes, those symbols are the chemical bases of DNA (or RNA, or perhaps even amino acids of proteins), and the “space” dimension is their position along some sequence.
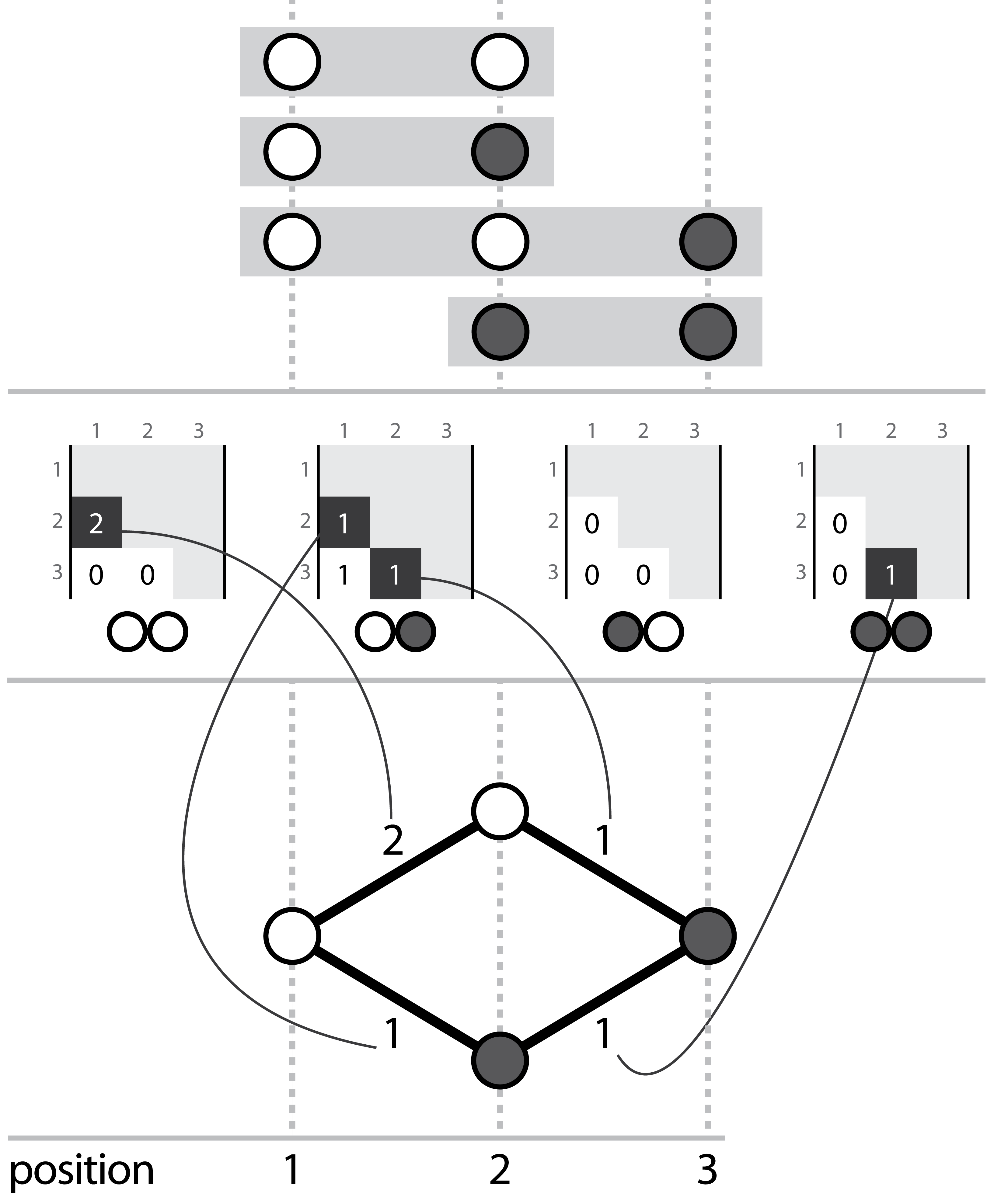
Three corresponding representations, (top) aligned reads, (middle) the Hansel structure, (bottom) a graph that can be derived from the Hansel structure
We fill this structure with counts of the number of times we observe some pair of nucleotides, at some pair of positions on the same read. Hansel provides a numpy ndarray backed class that offers a user friendly API for operating on our data structure: including adding, adjusting and fetching the counts of pairwise co-occurrences of pairs of symbols in space and time, and making probabilistic queries on the likelihood of symbols occurring, given some sequence of observed symbols thus far.
Gretel
Gretel is a Python package that now houses the actual algorithm that exploits the spun-out API offered by Hansel to attempt to recover haplotypes from the metagenome. Gretel provides a command line based tool that accepts a BAM of aligned reads (against a metagenomic pseudo-reference, typically the assembly) and a VCF of single nucleotide polymorphisms.
Gretel populates the Hansel matrix with observations by parsing the reads in the BAM at the genomic positions described in the provided VCF. Once parsing the reads is complete, Gretel exploits the ability to traverse the Hansel structure like a graph, creating chains of nucleotides that represent the most likely sequence of SNPs that appear on reads that in turn align to some region of interest on the pseudo-reference.
At each node, the decision to move to the next node (i.e. nucleotide) is made by ranking each of the probabilities of the node on the end of each current outgoing edge appearing after some subset of the previously seen nodes (i.e. the current path). Thus both the availability and associated probabilities of future edges are dependent on not only the current or previous node, but the path itself.
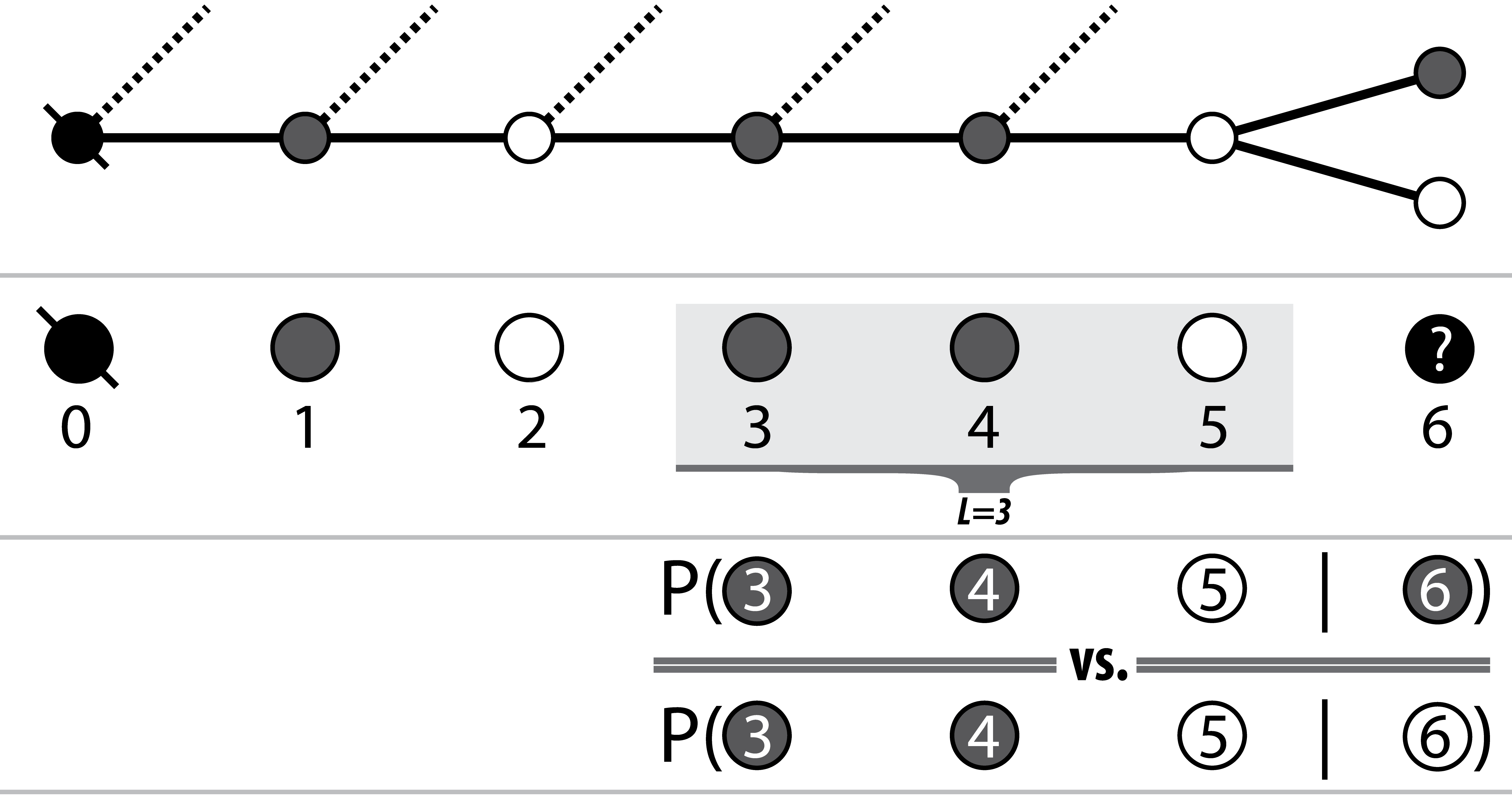
Pairwise conditionals between L last variants on the observed path,
and each of the possible next variants are calculated and the
best option (highest likelihood) is chosen
Gretel will construct a path in this way until a dummy sentinel representing the end of the graph is reached. After a path has been constructed from the dummy (or “sentinel”) start node to the end, observations in the Hansel structure are reweighted; to ensure that the same path is not returned again (as traversal is deterministic) and to allow Gretel to return the next most likely haplotype instead.
Gretel repeatedly attempts to traverse the graph, each time returning the next most likely haplotype given the reweighted Hansel structure, until a node with no outgoing edges is encountered (i.e. all observations between two genomic positions have been culled by re-weighting).
Going Public
After surgically separating Hansel from Gretel, the codebases were finally in a state where I wasn’t sick at the thought of anyone looking at them. Thus Hansel and Gretel now have homes on Github: see github.com/samstudio8/hansel and github.com/samstudio8/gretel. In an attempt to not be an awful person, partial documentation now also exists on ReadTheDocs: see hansel.readthedocs.io and gretel.readthedocs.io. Hansel and Gretel are both open source and distributed under the MIT Licence.
Reweighting
For the time being, I am settled on the methodology for reweighting the Hansel matrix as described in February’s status report, with the only recent difference being that it is now implemented correctly. Given a path through the metahaplome found by Gretel (that is, a predicted haplotype), we consider the marginal distribution of each of the selected variants that compose that path, and select the smallest (i.e. the least likely variant to be selected across all SNPs). That probability is then used as a ratio to reduce the observations in the Hansel data structure for all pairs of variants on the path.
As I have mentioned in the past, reweighting requires care: an overly aggressive methodology would dismiss similar looking paths (haplotypes) that have shared variants, but an overly cautious algorithm would limit exploration of the metahaplome structure and inhibit recovery of real haplotypes.
Testing so far has indicated that out of the methods I have proposed, this is the most robust, striking a happy balance between exploration-potential and accuracy. At this time I’m happy to leave it as is and work on something else, before I break it horribly.
The “Lookback” Parameter
As the calculation of edge likelihoods depends on a subset of the current path, a keen reader might wonder how such a subset is defined. Your typical bioinformatics software author would likely leave this as an exercise:
-l, --lookback [mysteriously important non-optional integer that fundamentally changes how well this software works, correct selection is a dark art, never set to 0, good luck.]
Luckily for both bioinformatics and my viva, I refuse to inflict the field’s next k-mer size, and there exists a reasonable intuition for the selection of L: the number of nodes over which to lookback (from and including the head of the current path) when considering the next node (nucleotide of the sequence). My intuition is that sequenced read fragments cover some limited number of SNP sites. Thus there will be some L after which it is typically unlikely that pairs of SNPs will co-occur on the same read between the next variant node i+1 and the already observed node (i+1) - L.
I say typically here because in actuality, SNPs will not be uniformly distributed2 and as such the value of L according to this intuition is going to vary across the region of interest depending on both the read sizes, mate pair insert sizes, coverage and indeed the distribution of the SNPs themselves. We thus consider the average number of SNPs covered by all reads parsed from the BAM, and use this to set the value of L.
Smoothing
Smoothing is a component of Hansel that has flown under the radar in my previous discussions. This is likely because I have not given it a second thought since being Belgian last year. Smoothing attempts to awkwardly sidestep two potential issues, namely overfitting and ZeroDivisionError.
For the former, we want to avoid scenarios where variant sites with very low read coverage (and thus few informative observations) are assumed to be fully representative of the true variation. In this case, smoothing effectively inserts additional observations that were not actually present in the provided reads to attempt to make up for potentially unseen observations.
For the latter case, consider the conditional probability of symbol a at position i occurring, given some symbol b at position j. This is defined (before smoothing is applied) by the Hansel framework as:

If one were to query the Hansel API with some selection of i, b and j such that there are no reads spanning i that feature symbol b at position j, a ZeroDivisionError will be raised. This is undesirable and cannot be circumvented by simply “catching” the error and returning 0, as the inclusion of a probability of 0 in a sequence of products renders the entire sequence of probabilities as 0, too.
The current smoothing method is merely “add one smoothing”, which modifies the above equation to artificially insert one (more) observation for each possible combination of symbols a and b between i and j. This avoids division by zero as there will always be at least one valid observation. However I suspect that to truly address the former problem, a more sophisticated solution is necessary.
Fortunately, it appears that in practice, for metagenomes with reasonable coverage, the first problem falls away and smoothing has a negligible effect on evaluation of edge probabilities. Despite this, the method of smoothing employed is admittedly naive and future work could potentially benefit from replacement. It should be noted that the influence of smoothing has the potential to become particularly pronounced after significant reweighting of the Hansel matrix (i.e. when very few observations remain).
Evaluation
Avid readers will be aware that evaluation of this method has been a persistent thorn in my side since the very beginning. There are no metagenomic test data sets that offer raw sequence reads, an assembly of those reads and a set of all expected (or indeed, even just some) haplotypes for a gene known to truly exist in that metagenome. Clearly this makes evaluation of a method for enumerating the most likely haplotypes of a particular gene from sequenced metagenomic reads somewhat difficult.
You might remember from my last status report that generating my own test data sets for real genes was both convoluted, and fraught with inconsistencies that made it difficult to determine whether unrecovered variants were a result of my approach, or an artefact of the data itself. I decided to take a step back and consider a simpler form of the problem, in the hope that I could construct an adequate testing framework for future development, and provide an empirical proof that the approach works (as well as a platform to investigate the conditions under which it doesn’t).
Trivial Haplomes: Triviomes
To truly understand and diagnose the decisions my algorithm was making, I needed a source of reads that could be finely controlled and well-defined. I also needed the workflow to generate those reads to be free of uncontrolled external influences (i.e. reads dropped by alignment, difficult to predict SNP calling).
To accomplish this I created a script to construct trivial haplomes: sets of short, randomly generated haplotypes, each of the same fixed length. Every genomic position of such a trivial haplotype was considered to be a site of variation (i.e. a SNP). Tiny reads (of a configurable size range, set to 3-5bp for my work) are then constructed by sliding windows across each of the random haplotypes. Additional options exist to set a per-base error rate and “slip” the window (to reduce the quality of some read overlaps, decreasing the number of shared paired observations).
Although this appears to be grossly unrepresentative of the real problem at hand — what technology even generates reads of 3-5bp? Don’t forget, Hansel and Gretel are designed to work directly with SNPs. Aligned reads are only parsed at positions specified in the VCF (i.e. the list of called variants) and so real sequences collapse to a handful of SNPs anyway. The goal here is not so much about accurately emulating real reads with real variation and error, but to establish a framework for controlled performance testing under varying conditions (e.g. how do recovery rates change with respect to alignment rate, error rate, number of SNPs, haplotypes etc).
We must also isolate the generation of input files (alignments and variant lists) from external processes. That is, we cannot use established tools for read alignment and variant calling. Whilst the results of these processes are tractable and deterministic, they confound the testing of my triviomes due to their non-trivial behaviour. For example, my tiny reads have a known correct alignment: each read is yielded from some window with a defined start and end position on a given haplotype. However read aligners discard, clip and “incorrectly” align these tiny test reads3. My reads are no longer under my direct control.
In the same fashion, despite my intention that every genomic position of my trivial haplome is a SNP, established variant callers and their diploid assumptions can simply ignore, or warp the calling of tri- or tetra-alleleic positions.
Thus my script is responsible for generating a SAM file, describing the alignment of the generated reads against a reference that does not exist, and a VCF, which simply enumerates all genomic positions on the triviome as a potential variant.
It’s important to note here that for Gretel (and recovery in general) the actual content of the reference sequence is irrelevant: the job of the reference (or pseudo-reference, as I have taken to call it for metagenomes) is to provide a shared co-ordinate system for the sequenced reads via alignment. In this case, the co-ordinates of the reads are known (we generated them!) and so the process of alignment is redundant and the reference need not exist at all.
Indeed, this framework has proved valuable. A harness script now rapidly and repeatedly generates hundreds of triviomes. Each test creates a number of haplotypes, with some number of SNPs. The harness can also specify an error rate, and how to sample the reads from each generated haplotype (uniformly, exponentially etc.). The resulting read alignment and variant list is thrown at Hansel and Gretel as part of the harness. Haplotypes recovered and output by Gretel can then be compared to the randomly generated input sequences for accuracy by merely constructing a matrix of Hamming distances (i.e. how many bases do not match for each output-input pair?). This is simple, fast and pretty effective, even if I do say so myself.
Despite its simplicity, this framework forms a basis for testing the packages during development, as well as giving us a platform on which to investigate the influence on recovery rate that parameters such as read length, number of haplotypes, number of SNPs, error rate, alignment rate have. Neat!
Synthetic Metagenomes
Of course, this triviome stuff is all well and good, but we need to prove that recovery is also possible on real genomic data. So we still left executing the rather convoluted looking workflow that I left you with at towards the end of my last report?
On the surface, that appears to be the case. Indeed, we must still simulate reads from some set of related genes, align those reads to some pseudo-reference, call for SNPs on that alignment and use the alignment and those called SNPs as the input to Gretel to recover the original genes that we generated reads from in the first place. Of course, we must also compare the haplotypes returned by Gretel to the known genes to evaluate the results.
But actually, the difficulty in the existing workflow is in the evaluation. Currently we use an alignment step to determine where each input gene maps to the selected pseudo-reference. This alignment is independent from the alignment of generated reads to the pseudo-reference. The hit table that describes how each input gene maps to the master is actually parsed as part of the evaluation step. To compare the input sequences against the recovered haplotypes, we need to know which parts of the input sequence actually overlap the recovered sequences (which share the same co-ordinates as the pseudo-reference), and where the start and end of that overlapping region exists on that particular input. The hit table effectively allows us to convert the co-ordinates of the recovered haplotypes, to those of the input gene. We can then compare bases by position and count how many match (and how many don’t).
Unsurprisingly, this got messy quite quickly, and the situation was exacerbated by subtle disagreements between the alignments of genes to the reference with BLAST and reads to the reference with bowtie2. This caused quite a bit of pain and ultimately ended with me manually inspecting sequences and writing my own BLAST-style hit tables to correct the evaluation process.
One afternoon whilst staring at Tablet and pondering my life choices, I wondered why I was even comparing the input and output sequences in this way. We’re effectively performing a really poor local alignment search between the input and output sequences. Using an aligner such as BLAST to compare the recovered haplotypes to the input sequences seems to be a rather intuitive idea. So why don’t we just do an actual local alignment?
Without a good answer, I tore my haplotype evaluation logic out of Gretel and put it in a metaphorical skip. Now we’ve dramatically simplified the process for generating and evaluating data sets. Hooray.
A bunch of small data sets now exist at github.com/SamStudio8/gretel-test and a framework of questionable bash scripts make the creation of new data sets rather trivial. Wonderful.
Results
So does this all actually work? Without wanting to step on the toes of my next blog post, it seems to, yes.
Accuracy across the triviome harness in general, is very good. Trivial haplotypes with up to 250 SNPs can be recovered in full, even in haplomes consisting of reads from 10 distinct, randomly generated haplotypes. Unsurprisingly, we’ve confirmed that increasing the number of haplotypes, and the number of SNPs on those haplotypes makes recovery more difficult.
To investigate recovery from metahaplomes of real genes, I’ve followed my previous protocol: select some gene (I’ve chosen DHFR and AIMP1), use BLAST to locate five arbitrary but similar genes from the same family, break them into reads and feed the alignment and called SNPs to Gretel with the goal of recovering the original genes. For both the DHFR and AIMP1 data sets, it is possible to get recovery rates of 95-100% for genes that look similar to the psuedo-reference and 80+% for those that are more dissimilar.
The relationship between pseudo-reference similarity and haplotype recovery rates might appear discouraging at first, but after digging around for the reasoning behind this result, it turns out not to be Gretel‘s fault. Reads generated from sequences that have less identity to the pseudo-reference are less likely to align to that pseudo-reference, and are more likely to be discarded. bowtie2 denies Gretel access to critical evidence required to accurately recover these less similar haplotypes.
This finding echoes an overarching theme I have encountered with current genomic tools and pipelines: not only are our current protocols and software not suitable for the analysis of metagenomes, but their underlying assumptions of diploidy are actually detrimental to the analyses we are conducting.
Making a start on the new paper pic.twitter.com/dlDKrGKUy2
— Sam Nicholls (@samstudio8) August 10, 2016
Introducing our Preprint
My work on everything so far has culminated in the production of a preprint: Advances in the recovery of haplotypes from the metagenome. It’s quite humbling to see the sum total of around 18 months of my life summed into a document. Flicking through it a few months after it went online, I still get a warm fuzzy feeling: it looks like real science! I provide a proper definition of the metahaplome and introduce both the underlying graph theory for Hansel and the probability equations for Gretel. It also goes into a whole heap of results obtained so far, some historical background into the problem and where we are now as a field, and an insight into how this approach is different from other methodologies.
Our work is an advance in computational methods for extracting exciting exploitable enzymes from metagenomes.
Conclusion
Things appear to be going well. Next steps are to get in the lab and try Gretel out for real. We are still hunting around for some DNA in a freezer that has a corresponding set of good quality sequenced reads.
In terms of development for Hansel and Gretel, there is still plenty of room for future work (helpfully outlined by my preprint) but in particular I still need to come up with a good way to incorporate quality scores and paired end information. I expect both attributes will improve performance more than the pretty awesome results we are getting already.
For more heavy refactoring, I’d like to also look at replacing Gretel‘s inherent greedy bias (the best choice is always the edge with the highest probability) with something a little more clever. Additionally, the handling of indels is more than certainly going to become the next thorn in my side.
I’d also like to come up with some way of visualising results (I suspect it will involve Circos), because everybody can get behind a pretty graph.
In other news
I went in the lab for the first time…
AUTHORISED TO WORK IN A CLASS 2 GM LAB, GET READY FOR SCIENCE
— Sam Nicholls (@samstudio8) June 15, 2016
…it was not a success
LOOK AT THIS SHIT pic.twitter.com/jkdIVxeZVz
— Sam Nicholls (@samstudio8) June 22, 2016
Biologists on Reddit actually liked my sassy PCR protocol
omg the real biologists are talking to me like a NOT PRETEND BIOLOGISThttps://t.co/QDpneYoDBY pic.twitter.com/lzAb1TdLpX
— Sam Nicholls (@samstudio8) June 21, 2016
Our sys admin left
Trusty sysadmin couldn’t give root, but a nice dev machine. Here’s the MOTD. Thanks and good luck, @MartinjVickers pic.twitter.com/1YMXZxfcVL
— Sam Nicholls (@samstudio8) May 20, 2016
This happened.
SO WE AREN’T IN EUROPE AND NOW TGAC IS CALLED THE EARLHAM INSTITUTE I DON’T UNDERSTAND ANYTHING ANYMORE
— Sam Nicholls (@samstudio8) June 27, 2016
Someone is actually using Goldilocks
SOMEONE IS USING MY SOFTWARE :’) https://t.co/tBo02HfyL0
— Sam Nicholls (@samstudio8) July 7, 2016
I am accidentally in charge of the department 3D printer
YESSSSSS IT’S HERE pic.twitter.com/NjO0afZcgA
— Sam Nicholls (@samstudio8) July 12, 2016
Writing a best man’s speech is definitely harder than an academic paper
I WAS AT A WEDDING pic.twitter.com/t36bW1XIE6
— Sam Nicholls (@samstudio8) July 18, 2016
I am a proper biologist now
I FEAR I AM NOT COMPETITIVE ENOUGH FOR MY ENVIRONMENT, I AM OVERRUN, SEND HELP HUMANS pic.twitter.com/IQF078qxud
— Sam Nicholls (@samstudio8) July 27, 2016
Someone trusts me to play with lasers
PEW PEW pic.twitter.com/JEXsoffBFm
— Sam Nicholls (@samstudio8) August 3, 2016
I acquired some bees
AWAITING DUMPER TRUCK OF BEES #BEES @DarkestKale pic.twitter.com/s5xEr1aexT
— Sam Nicholls (@samstudio8) August 6, 2016
I went up some mountains with a nice man from the internet
Mountain 1 complete pic.twitter.com/9CvgSsmCwg
— Sam Nicholls (@samstudio8) August 13, 2016
I went to a conference…
At the @eccb2016 welcome party. The drinks don't appear to be limited, so I'm on my fifth wine. Cheers :3 pic.twitter.com/QBTjwmIMYY
— Sam Nicholls (@samstudio8) September 4, 2016
…I presented some work!
Visit DA125 to hear how to pull exciting enzymes out of metagenomes before I have too much free wine #eccb2016 pic.twitter.com/7o5ueuXLnS
— Sam Nicholls (@samstudio8) September 5, 2016
I accidentally went on holiday
Me looking at email on holiday pic.twitter.com/5FLWZamsnZ
— Sam Nicholls (@samstudio8) September 12, 2016
…and now I am back.
Urgh. pic.twitter.com/vzRZmq7xuw
— Sam Nicholls (@samstudio8) September 28, 2016
tl;dr
- Yes hello, I am still here and appear to be worse at both blog and unit testing
- My work now has a name: a data structure called
Hanseland an algorithm calledGretel - Reweighting appears to work in a robust fashion now that it is implemented correctly
- Smoothing the conditional probabilities of observations should be looked at again
- Triviomes provide a framework for investigating the effects of various aspects of metagenomes on the success of haplotype recovery
- Generating synthetic metahaplomes for testing is significantly less of a pain in the ass now I have simplified the process for evaluating them
- Current protocols and software are not suitable for the analysis of metagenomes (apart from mine)
- I have a preprint!
- Indels are almost certainly going to be the next pain in my ass
- I shunned my responsibilities as a PhD student for a month and travelled Eastern Europe enjoying the sun and telling strangers about how awful bioinformatics is
- I am now back telling North Western Europeans how awful bioinformatics is
- Mhaplotyper? Oh yes, the M is for metagenomic, and it is also silent. ↩
-
Having written this paragraph, I wonder what the real impact of this intuition actually is. I’ve now opened an issue on my own repo to track my investigation. In the case where more than
Lpairs of evidence exist, is there quantifiable loss in accuracy by only consideringLpairs of evidence? In the case where fewer thanLpairs of evidence exist, does the smoothing have a non-negligible affect on performance? ↩ - Of course, the concept of alignment in a triviome is somewhat undefined as we have no “reference” sequence anyway. Although one could select one of the random haplotypes as a pseudo-reference against which to align all tiny reads, considering the very short read lengths and the low sequence identity between the randomly generated haplotype sequences, it is highly likely that the majority of reads will fail to align correctly (if at all) to such a reference. ↩