Yesterday I spoke at the Centre of Computational Biology at Birmingham University. I was invited to give a talk as part of their research seminar series about the work I have been doing on metagenomes. The lead up to this has been pretty nerve-wracking as this was my first talk outside of Aberystwyth (since my short introductory talk at KU Leuven last year), and the majority of my previous talks have been to my peers, which I find to be a lot less intimidating than a room full of experts of various fields.
En route to @UoB_CCB to shout about why metagenomes are so hard pic.twitter.com/wftx6sUhWV
— Sam Nicholls (@samstudio8) November 2, 2016
Metaphorical Metagenomes
I submitted the current working title of my PhD: “Extracting exciting exploitable enzymes from massive metagenomes“, which I think is a rather catchy summary of what I’m working on here. I borrowed the opening slides from my previous talks (this is a cow…) but felt like I needed to try a new explanation of why the metagenome is so difficult to work with. Previously, I’ve described the problem with jigsaw puzzles: i.e. consider many distinct (but visually similar) jigsaws, mixed together (with duplicate and missing pieces). Whilst this is a nice, accessible description that appears to serve well, it tends to leave some listeners confused about my objective, particularly:
- You are recovering whole genomes?
The jigsaw metaphor doesn’t lend well to the metahaplome and the concept of assembling a jigsaw partially. Listeners assume we want to piece together each of the different jigsaws in our box, whole – presumably because people find those who don’t finish jigsaws terrible. - We can assemble jigsaws incorrectly?
Metagenomic assemblies are a consensus sequence of the observed reads. The resulting sequence is unlikely to exist in nature. Whilst we can extend our metaphor to explain that pieces of jigsaws may have the same shape, such that we can put together puzzles that don’t exist, this is not immediately obvious to listeners.
A common analogy for genomic assembly is that of pages shredded from a book. I’ve also previously pitched this at a talk to try and explain metagenomic assembly, but this has some disastrous metaphorical pitfalls too:
- You are recovering whole books?
Akin to the jigsaw analogy, listeners don’t immediately see why we would only want to assemble parts of a book. What part? A chapter? A page? A paragraph? Which leads to… - Why are there paragraphs shared between books?
To describe our problem of recovering genes that appear across multiple species, we must say that we are attempting to recover some shared sequence of words from across many books. This somewhat breaks the metaphor as this isn’t a problem that exists, and so the concept just causes listener confusion, rather than helping them to understand our problem. Whilst we could point out the Bible as an example of a book that has been translated and shared to a point where two copies of the text do feature differences between their passages, we figure it best to avoid conversations about the Bible and shredded books. - You are assembling words into sentences? The problem is easy?
DNA has a limited alphabet: A, C, G and T. But books can contain a practically infinite combination of character sequences given their larger alphabets. This larger alphabet makes distinguishing sequence similarity incredibly simple compared to that of DNA. Right now I’m using an alphabet of about 95 characters (upper and lowercase characters, numbers and a subset of symbols) in this post, and although it’s possible that one or more of my sentences could appear elsewhere on the web (unintentionally), the probability of this will be many, many orders of magnitude smaller than that of finding duplication of DNA sequences within and between genomes. Thus by comparing the problem to reconstructing pages from a book, we are at a very real risk of underselling the difficulty of the problem at hand.
Additionally, both analogies fail to explain:
- Intra-object variation
We must also shoehorn the concept of intraspecies gene variation into these metaphors which turns out rather clunky. We do say that books and jigsaws have misprints and errors, but this doesn’t truly emphasise that there is real variation between instances of the same object. - What is the biological significance anyway?
Neither description of the problem comes close to explaining why we’d even want to retrieve the different but similar-looking areas of a jigsaw, or copies of a page or passage shared across multiple books.
Machines and Factories: A new metaphor?
So, I spent some time iterating over many ideas and came up with a new concept of “genes as machines” and “genomes as factories”:
Genes
Consider a gene as a physical machine. It’s configuration is set by altering the state of its switches. The configuration of a machine is akin to a sequence of DNA. It is possible (and even intended) that the machine can be configured in many different ways by changing the state of its switches (like gene variants), but it is still the same machine (the same gene). This is an important concept because we want to describe that a machine can have many configurations (that can do nothing, improve performance, or even break it), whilst still remaining the same machine (i.e. a variant of a gene).

Factories
We can consider a genome as a factory, holding a collection of machines and their configurations:
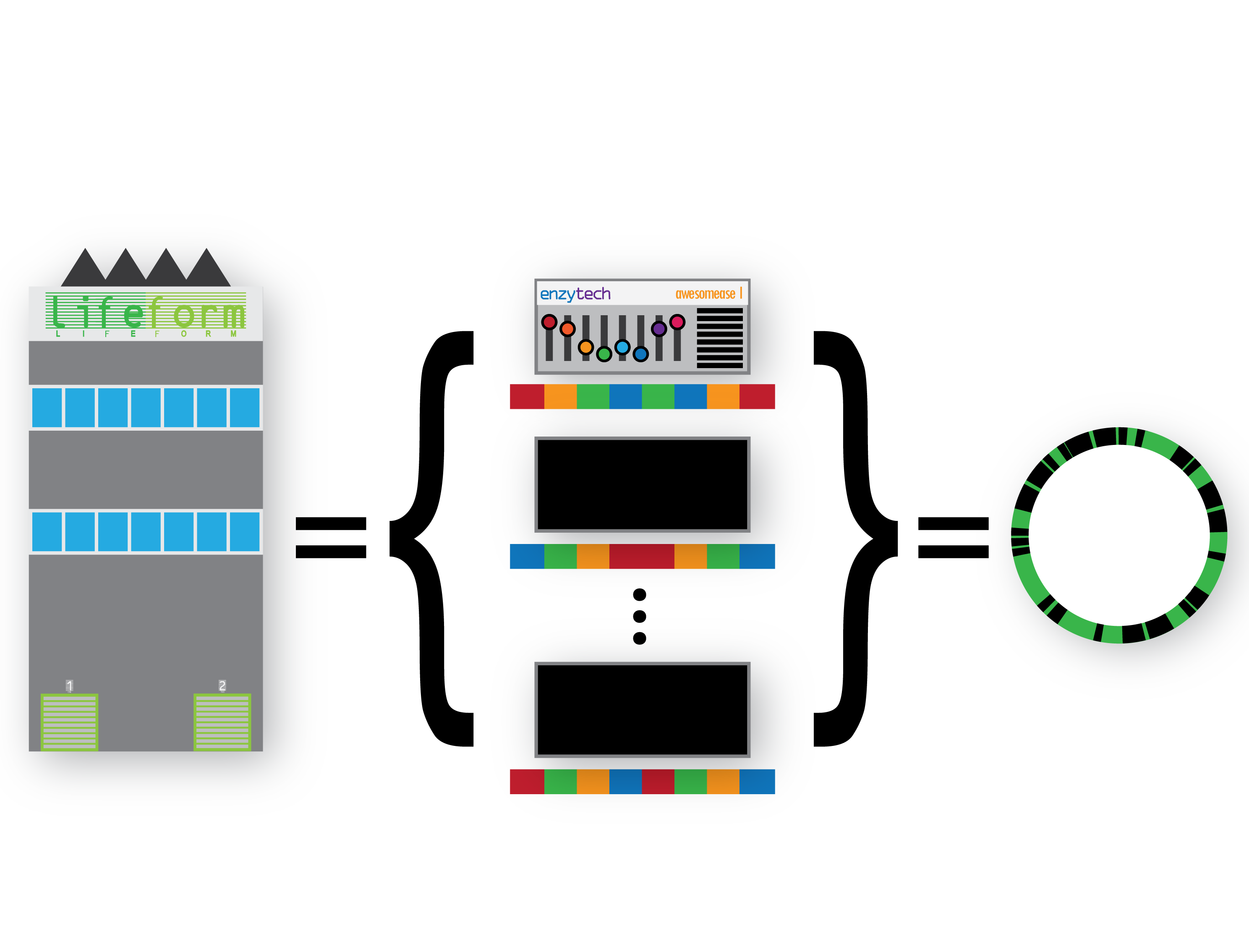
We can extend this metaphor to groups of factories under a parent organisation (i.e. a species) who can set the configuration of their machines autonomously – introducing intra-species variation as a concept. Additionally we can describe groups of factories under other parent organisations (species) that also deploy the same machine to their own factories, also configuring them differently – introducing not only intra-species variation, but multiple sets of intra-species variants too:
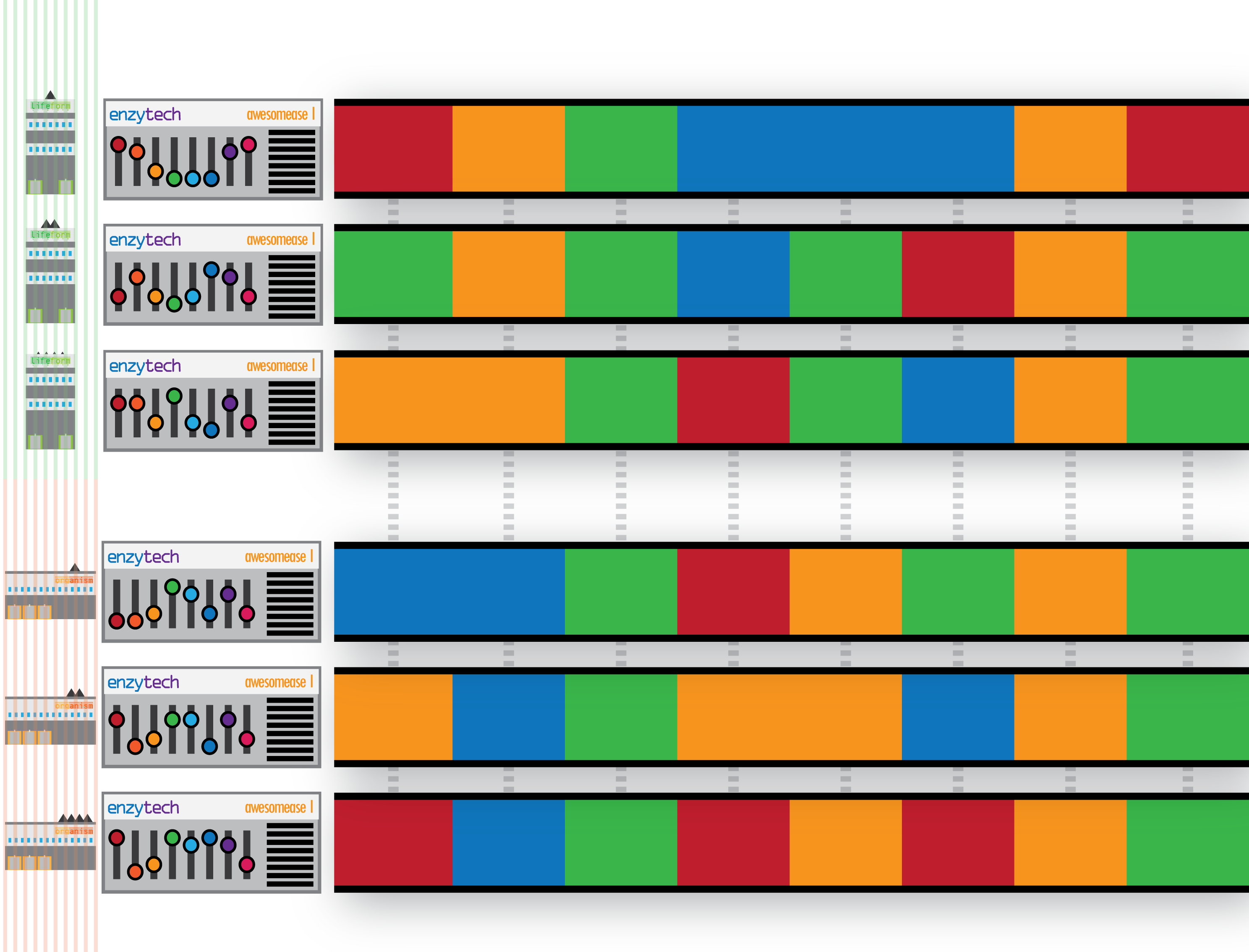
Talk the Talk
Armed with my shiny diagrams and apprehension of my own new metaphor, I pitched it to my listeners as a test and thanked them for their role as guinea pigs to my new attempt at explaining the core concept of the metagenome and its problems.
PREPARE YOURSELVES pic.twitter.com/s1fcccXNgQ
— Sam Nicholls (@samstudio8) November 2, 2016
In general, I felt like the audience followed along with the metaphor to begin with. Given a fictional company: Enzytech and their machine: Awesomease, we could define the metahaplome as the collection of configurations of that Enzytech Awesomease product across multiple factories, under various parent companes (i.e. different genomes, of varying species). However I think the story unravelled when I described the process of actually recovering the metahaplome.
I set the scene: Sam from Enzytech wondered why factories configured their Awesomease differently. Sam figured there must be an interesting meaning to these configurations – do some combinations of switches cause the Awesomease to -ase more awesomely? Thus, Sam approaches each parent company and requests their Enzytech Awesomease configurations. In a surprising gesture of co-operation, the businesses comply and return all their Enzytech Awesomease configurations, for all of their factories. Unfortunately, and perhaps in breach of their own trade secrets, they also submit the configurations of every other machine (gene) in each of their factories (genomes) too:
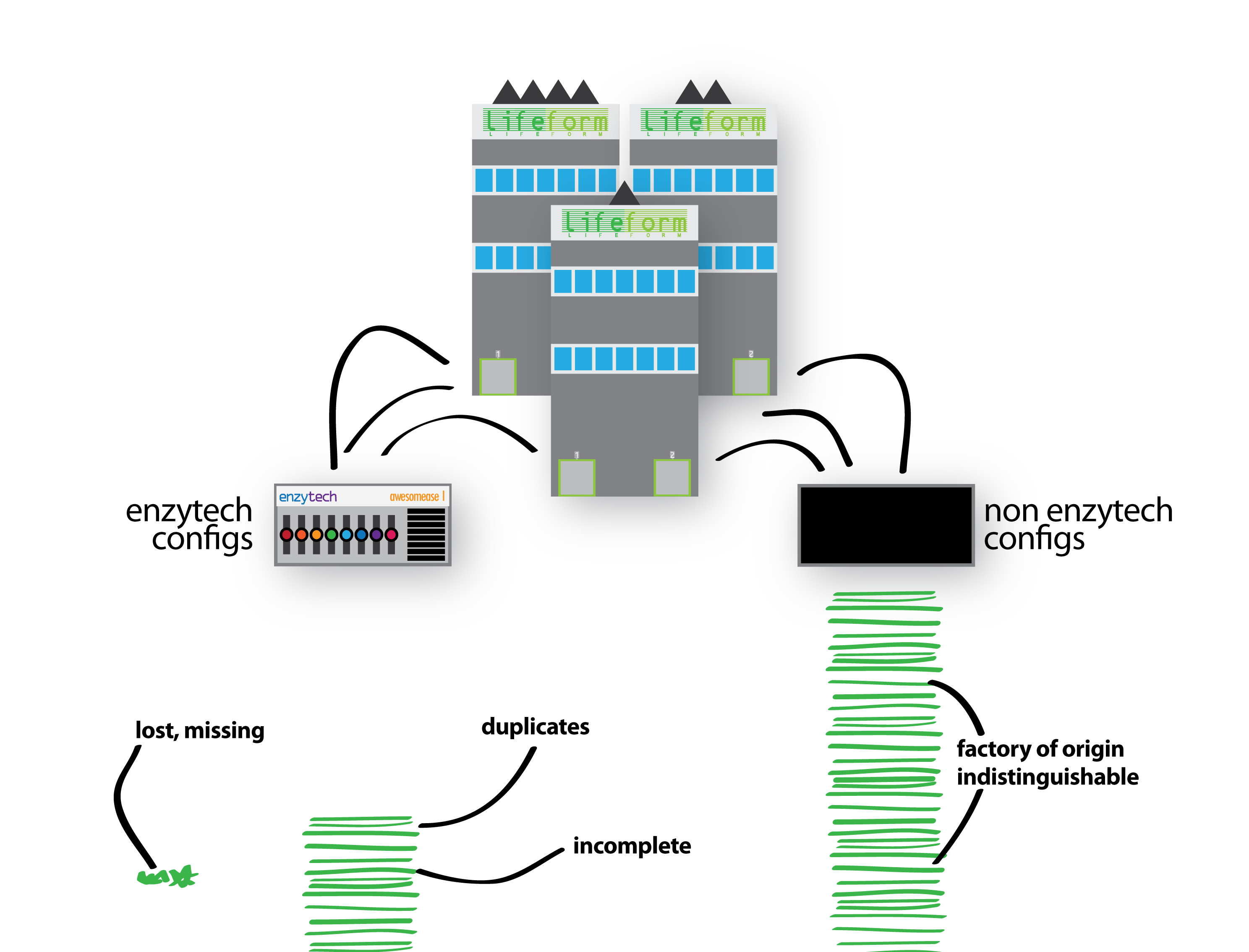
To make matters worse, the configurations don’t describe the specific factory they are from (i.e. the individual organism), and their returned documents also include incomplete, broken and duplicated configurations. Lost configurations are not submitted.
I think at this point, I was getting too wrapped up in the metaphor and its story. The concept of metaphorical factories submitting bad paperwork to fictional Sam from Enzytech did not have an obvious biological reference point (it was supposed to describe metagenomic sampling). I think with practice, I could deliver this part better such that my audience understands the relevance to biology, but I am not sure it is necessary. Where things definitely did not work was this slide:
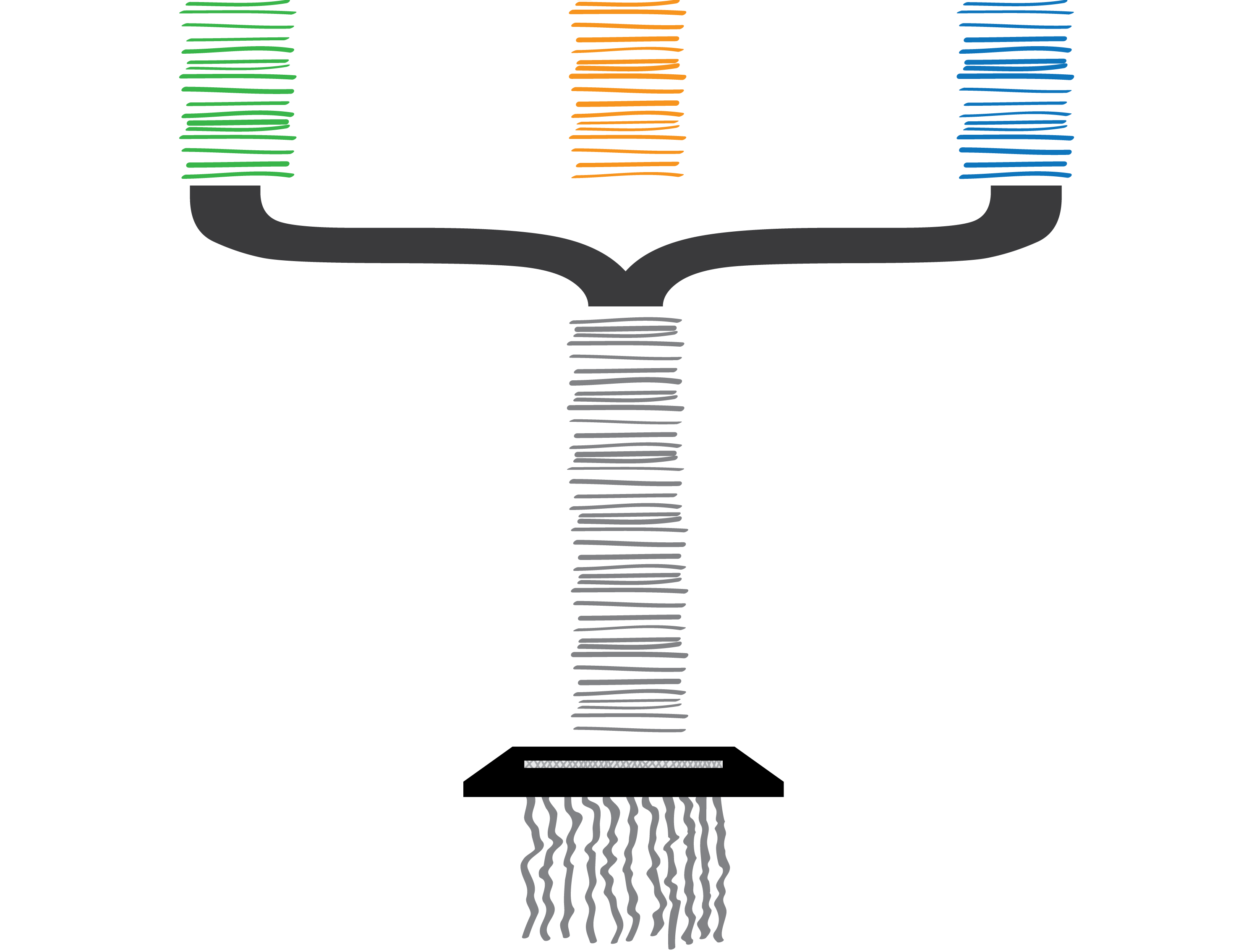
“Unfortunately, an Enzytech intern misfiled the paperwork submitted by all of the parent companies’ factories (species and their various genomes), and we could no longer determine which company submitted which configuration. The same clumsy intern then accidentally shredded all of the configurations, too.”
Welp. I am somewhat cringing at the amount of biological shoehorning going on in just one slide. Here I wanted to explain that although my pretty pictures have helpful colour coding for each of the companies (species), we don’t have this in our data. A configuration (gene variant) could come from any factory (genome) in our sample, and there is no way of differentiating them. Although shredding is a (common) reference to short-read sequencing technology, the delivery of this slide feels as clumsy as the Enzytech intern. I think the mistake I have made here was trying to use the same metaphorical explanation for two separate and distinct problems that I face in my work on metagenomes:
- The metahaplome
We need to clearly define what the metahaplome actually is as it is a term we coined. It is also the objective of my algorithm, and so failing to adequately describe this means it is unclear why this work has a biological relevance (or is worth a PhD). - Metagenomes, assembly, and short read sequencing
This final slide attempts to describe metagenomes and sequencing, as shredded paperwork relating to many different genes, from multiple factories that are held by various parent companies, all mixed together. But in fact, for this part of the metaphor it is easier to just say “bits of DNA, from a gene, on multiple organisms, from multiple species in the same environmental sample”…
On this occasion, I believe I managed to explain the metahaplome more clearly to an audience than ever before, though this might be in part because this is my first talk since our pre-print. However, in forcing my new metaphor onto the latter problem (of sequencing), I think I inadvertently convoluted what the metagenome is. So ultimately, I’m not entirely convinced the new metaphor panned out with a mixed audience of expert computer scientists and biologists. That said, I had several excellent questions following the talk, that seemed to imply a deep understanding of the work I presented, so hooray! Regardless of whether I deploy it for my next talk, I think it will still prove a nice way to explain my work to the public at large (who may have no frame of reference to get confused with).
I enjoyed the opportunity to visit somewhere new and speak about my work, especially as my first invited talk outside of Aberystwyth. This is also a reminder that even sharing thoughts and progress on cross-discipline work is hard. It’s a lot of work to come up with a way to get everyone in the audience on the same page; capable of speaking the same language (biological, mathematical and computational terminology) and also give the necessary background knowledge (genomic sequencing and metagenomes) to even begin to pitch the novelty and importance of our own work.
Obligatory proof that people attended:
— CCB UoB (@UoB_CCB) November 2, 2016
Obligatory omg my heart rate:
tl;dr
- I was invited to speak at Birmingham, it was nice
- It’s super hard to come up with explanations of your work that will please everyone
- Spending until 4am drawing some rather shiny diagrams is perhaps not the best reason to push forth with a new metaphor that even you feel a little uneasy about
- I continue to speak too bloody quickly
- My body still gives the physiological impression I am doing exercise whilst speaking publicly