chitin is.Back in 2014, about half way through my undergraduate dissertation (Application of Machine Learning Techniques to Next Generation Sequencing Quality Control), I made an unsettling discovery.
I am disorganised.
The discovery was made after my supervisor asked a few interesting questions regarding some of my earlier discarded analyses. When I returned to the data to try and answer those questions, I found I simply could not regenerate the results. Despite the fact that both the code and each “experiment” were tracked by a git repository and I’d written my programs to output (what I thought to be) reasonable logs, I still could not reproduce my science. It could have been anything: an ad-hoc, temporary tweak to a harness script, a bug fix in the code itself masking a result, or any number of other possible untracked changes to the inputs or program parameters. In general, it was clear that I had failed to collect all pertinent metadata for an experiment.
Whilst it perhaps sounds like I was guilty of negligent book-keeping, it really wasn’t for lack of trying. Yet when dealing with many interesting questions at once, it’s so easy to make ad-hoc changes, or perform undocumented command line based munging of input data, or accidentally run a new experiment that clobbers something. Occasionally, one just forgets to make a note of something, or assumes a change is temporary but for one reason or another, the change becomes permanent without explanation. These subtle pipeline alterations are easily made all the time, and can silently invalidate swathes of results generated before (and/or after) them.
Ultimately, for the purpose of reproducibility, almost everything (copies of inputs, outputs, logs, configurations) was dumped and tar‘d for each experiment. But this approach brought problems of its own: just tabulating results was difficult in its own right. In the end, I was pleased with that dissertation, but a small part of me still hurts when I think back to the problem of archiving and analysing those result sets.
It was a nightmare, and I promised it would never happen again.
Except it has.
A relapse of disorganisation
Two years later and I’ve continued to be capable of convincing a committee to allow me to progress towards adding the title of doctor to my bank account. As part of this quest, recently I was inspecting the results of a harness script responsible for generating trivial haplotypes, corresponding reads and attempting to recover them using Gretel. “Very interesting, but what will happen if I change the simulated read size”, I pondered; shortly before making an ad-hoc change to the harness script and inadvertently destroying the integrity of the results I had just finished inspecting by clobbering the input alignment file used as a parameter to Gretel.
Argh, not again.
Why is this hard?
Consider Gretel: she’s not just a simple standalone tool that one can execute to rescue haplotypes from the metagenome. One must go through the motions of pushing their raw reads through some form of pipeline (pictured below) to generate an alignment (to essentially give a co-ordinate system to those reads) and discover the variants (the positions in that co-ordinate system that relate to polymorphisms on reads) that form the required inputs for the recovery algorithm first.
This is problematic for one who wishes to be aware of the providence of all outputs of Gretel, as those outputs depend not only on the immediate inputs (the alignment and called variants), but the entirety of the pipeline that produced them. Thus we must capture as much information as possible regarding all of the steps that occur from the moment the raw reads hit the disk, up to Gretel finishing with extracted haplotypes.
But as I described in my last status report, these tools are themselves non-trivial. bowtie2 has more switches than an average spaceship, and its output depends on its complex set of parameters and inputs (that also have dependencies on previous commands), too.
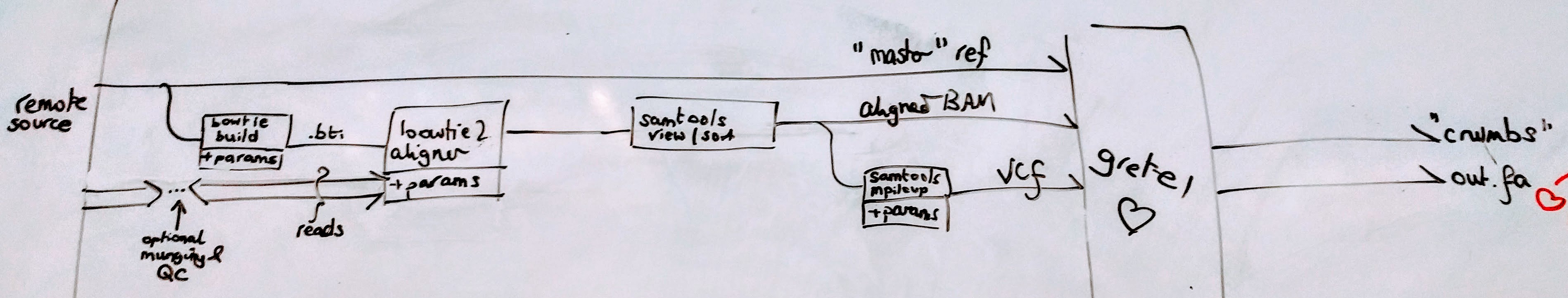
bash scripts are all well and good for keeping track of a series of commands that yield the result of an experiment, and one can create a nice new directory in which to place such a result at the end – along with any log files and a copy of the harness script itself for good measure. But what happens when future experiments use different pipeline components, with different parameters, or we alter the generation of log files to make way for other metadata? What’s a good directory naming strategy for archiving results anyway? What if parts (or even all of the) analysis are ad-hoc and we are left to reconstruct the history? How many times have you made a manual edit to a malformed file, or had to look up exactly what combination of sed, awk and grep munging you did that one time?
One would have expected me to have learned my lesson by now, but I think meticulous digital lab book-keeping is just not that easy.
What does organisation even mean anyway?
I think the problem is perhaps exacerbated by conflating the meaning of “organisation”. There are a few somewhat different, but ultimately overlapping problems here:
- How to keep track of how files are created
What command created filefoo? What were the parameters? When was it executed, by whom? - Be aware of the role that each file plays in your pipeline
What commands go on to use filefoo? Is it still needed? - Assure the ongoing integrity of past and future results
Does this alignment have reads? Is that FASTA index up to date?
Are we about to clobber shared inputs (large BAMS, references) that results depend on? - Archiving results in a sensible fashion for future recall and comparison
How can we make it easy to find and analyse results in future?
Indeed, my previous attempts at organisation address some but not all of these points, which is likely the source of my bad feeling. Keeping hold of bash scripts can help me determine how files are created, and the role those files go on to play in the pipeline; but results are merely dumped in a directory. Such directories are created with good intent, and named something that was likely useful and meaningful at the time. Unfortunately, I find that these directories become less and less useful as archive labels as time goes on… For example, what the fuck is ../5-virus-mix/2016-10-11__ref896__reg2084-5083__sd100/1?
This approach also had no way to assure the current and future integrity of my results. Last month I had an issue with Gretel outputting bizarrely formatted haplotype FASTAs. After chasing my tail trying to find a bug in my FASTA I/O handling, I discovered this was actually caused by an out of date FASTA index (.fai) on the master reference. At some point I’d exchanged one FASTA for another, assuming that the index would be regenerated automatically. It wasn’t. Thus the integrity of experiments using that combination of FASTA+index was damaged. Additionally, the integrity of the results generated using the old FASTA were now also damaged: I’d clobbered the old master input.
There is a clear need to keep better metadata for files, executed commands and results, beyond just tracking everything with git. We need a better way to document the changes a command makes in the file system, and a mechanism to better assure integrity. Finally we need a method to archive experimental results in a more friendly way than a time-sensitive graveyard of timestamps, acronyms and abbreviations.
So I’ve taken it upon myself to get distracted from my PhD to embark on a new adventure to save myself from ruining my PhD2, and fix bioinformatics for everyone.
Approaches for automated command collection
Taking the number of post-its attached to my computer and my sporadically used notebooks as evidence enough to outright skip over the suggestion of a paper based solution to these problems, I see two schools of thought for capturing commands and metadata computationally:
- Intrusive, but data is structured with perfect recall
A method whereby users must execute commands via some sort of wrapper. All commands must have some form of template that describes inputs, parameters and outputs. The wrapper then “fills in” the options and dispatches the command on the user’s behalf. All captured metadata has uniform structure and nicely avoids the need to attempt to parse user input. Command reconstruction is perfect but usage is arguably clunky. - Unobtrusive, best-effort data collection
A daemon-like tool that attempts to collect executed commands from the user’s shell and monitor directories for file activity. Parsing command parameters and inputs is done in a naive best-effort scenario. The context of parsed commands and parameters is unknown; we don’t know what a particular command does, and cannot immediately discern between inputs, outputs, flags and arguments. But, despite the lack of structured data, the user does not notice our presence.
There is a trade-off between usability and data quality here. If we sit between a user and all of their commands, offering a uniform interface to execute any piece of software, we can obtain perfectly structured information and are explicitly aware of parameter selections and the paths of all inputs and desired outputs. We know exactly where to monitor for file system changes, and can offer user interfaces that not only merely enumerate command executions, but offer searching and filtering capabilities based on captured parameters: “Show me assemblies that used a k-mer size of 31”.
But we must ask ourselves, how much is that fine-grained data worth to us? Is exchanging our ability to execute commands ourselves worth the perfectly structured data we can get via the wrapper? How much of those parameters are actually useful? Will I ever need to find all my bowtie2 alignments that used 16 threads? There are other concerns here too: templates that define a job specification must be maintained. Someone must be responsible for adding new (or removing old) parameters to these templates when tools are updated. What if somebody happens to misconfigure such a template? More advanced users may be frustrated at being unable to merely execute their job on the command line. Less advanced users could be upset that they can’t just copy and paste commands from the manual or biostars. What about smaller jobs? Must one really define a command template to run trivial tools like awk, sed, tail, or samtools sort through the wrapper?
It turns out I know the answer to this already: the trade-off is not worth it.
Intrusive wrappers don’t work: a sidenote on sunblock
Without wanting to bloat this post unnecessarily, I want to briefly discuss a tool I’ve written previously, but first I must set the scene3.
Within weeks of starting my PhD, I made a computational enemy in the form of Sun Grid Engine: the scheduler software responsible for queuing, dispatching, executing and reporting on jobs submitted to the institute’s cluster. I rapidly became frustrated with having an unorganised collection of job scripts, with ad-hoc edits that meant I could no longer re-run a job previously executed with the same submission script (does this problem sound familiar?). In particular, I was upset with the state of the tools provided by SGE for reporting on the status of jobs.
To cheer myself up, I authored a tool called sunblock, with the goal of never having to look at any component of Sun Grid Engine directly ever again. I was successful in my endeavour and to this day continue to use the tool on the occasion where I need to use the cluster.
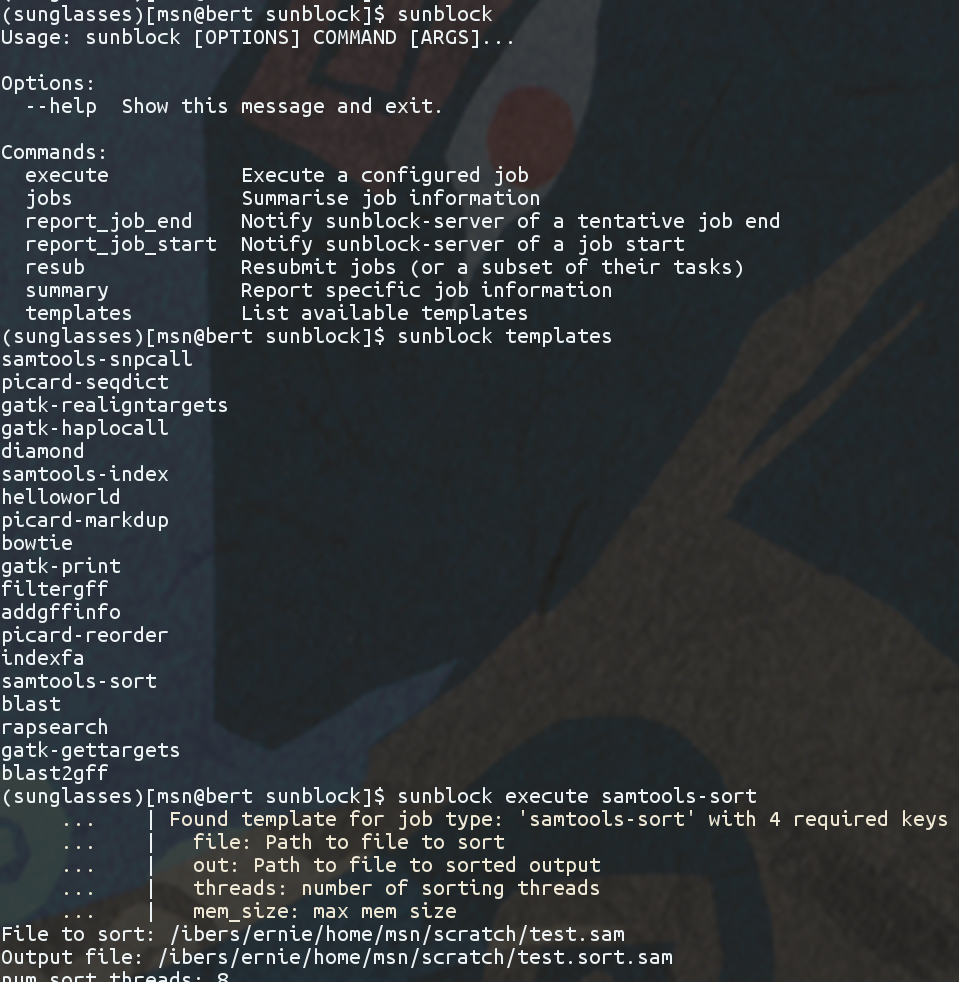
However, as hypothesised above, sunblock does indeed require an explicit description of an interface for any job that one would wish to submit to the cluster, and it does prevent users from just pasting commands into their terminal. This all-encompassing wrapping feature; that allows us to capture the best, structured information on every job, is also the tool’s complete downfall. Despite the useful information that could be extracted using sunblock (there is even a shiny sunblock web interface), its ability to automatically re-run jobs and the superior reporting on job progress compared to SGE alone, was still not enough to get user traction in our institute.
For the same reason that I think more in-the-know bioinformaticians don’t want to use Galaxy, sunblock failed: because it gets in the way.
Introducing chitin: an awful shell for awful bioinformaticians
Taking what I learned from my experimentation with sunblock on-board, I elected to take the less intrusive, best-effort route to collecting user commands and file system changes. Thus I introduce chitin: a Python based tool that (somewhat)-unobtrusively wraps your system shell, to keep track of commands and file manipulations to address the problem of not knowing how any of the files in your ridiculously complicated bioinformatics pipeline came to be.
I initially began the project with a view to create a digital lab book manager. I envisaged offering a command line tool with several subcommands, one of which could take a command for execution. However as soon as I tried out my prototype and found myself prepending the majority of my commands with lab execute, I wondered whether I could do better. What if I just wrapped the system shell and captured all entered commands? This might seem a rather dumb and long-about way of getting one’s command history, but consider this: if we wrap the system shell as a means to capture all the input, we are also in a position to capture the output for clever things, too. Imagine a shell that could parse the stdout for useful metadata to tag files with…
I liked what I was imagining, and so despite my best efforts to get even just one person to convince me otherwise; I wrote my own pseudo-shell.
WHAT'S THAT? A SHELL WITH BUILT IN FUNCTIONS FOR HOW YOUR FILES HAPPENED AND WHAT YOU NEED TO REPEAT TO GET TO A GIVEN FILE? WHY YES IT IS pic.twitter.com/h87pzptq1E
— Sam Nicholls (@samstudio8) November 15, 2016
chitin is already able to track executed commands that yield changes to the file system. For each file in the chitin tree, there is a full modification history. Better yet, you can ask what series of commands need to be executed in order to recreate a particular file in your workflow. It’s also possible to tag files with potentially useful metadata, and so chitin takes advantage of this by adding the runtime4, and current user to all executed commands for you.
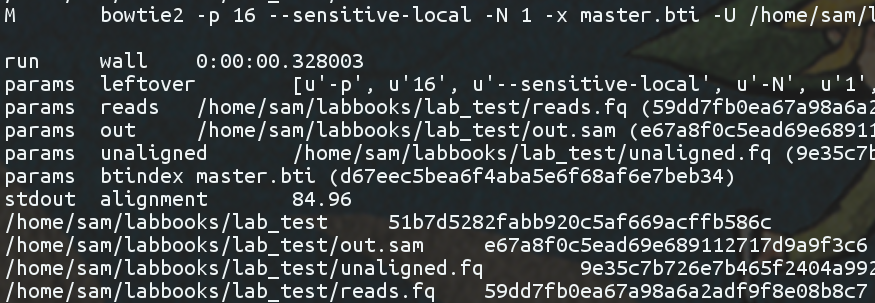
Additionally, I’ve tried to find my own middle ground between the sunblock-esque configurations that yielded superior metadata, and not getting in the way of our users too much. So one may optionally specify handlers that can be applied to detected commands, and captured stdout/stderr. For example, thanks to my bowtie2 configuration, chitin tags my out.sam files with the overall alignment rate (and a few targeted parameters of interest), automatically.
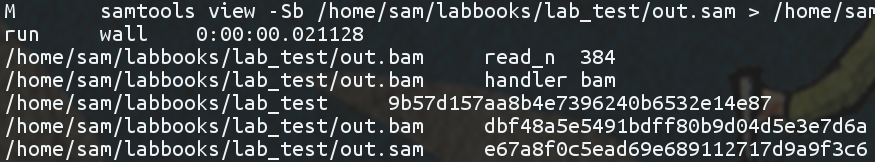
chitin also allows you to specify handlers for particular file formats to be applied to files as they are encountered. My environment, for example, is set-up to count the number of reads inside a BAM, and associate that metadata with that version of the file:
In this vein, we are in a nice position to check on the status of files before and after a command is executed. To address some of my integrity woes, chitin allows you to define integrity handlers for particular file formats too. Thus my environment warns me if a BAM has 0 reads, is missing an index, or has an index older than itself. Similarly, an empty VCF raises a warning, as does an out of date FASTA index. Coming shortly will be additional checks for whether you are about to clobber a file that is depended on by other files in your workflow. Kinda cool, even if I do say so myself.
Conclusion
Perhaps I’m trying to solve a problem of my own creation. Yet from a few conversations I’ve had with folks in my lab, and frankly, anyone I could get to listen to me for five minutes about managing bioinformatics pipelines, there seems to be sympathy to my cause. I’m not entirely convinced myself that a “shell” is the correct solution here, but it does seem to place us in the best position to get commands entered by the user, with the added bonus of getting stdout to parse for free. Though, judging by the flurry of Twitter activity on my dramatically posted chitin screenshots lately, I suspect I am not so alone in my disorganisation and there are at least a handful of bioinformaticians out there who think a shell isn’t the most terrible solution to this either. Perhaps I just need to be more of a wet-lab biologist.
Either way, I genuinely think there’s a lot of room to do cool stuff here, and to my surprise, I’m genuinely finding chitin quite useful already. If you’d like to try it out, the source for chitin is open and free on GitHub. Please don’t expect too much in the way of stability, though.
tl;dr
- A definition of “being organised” for science and experimentation is hard to pin down
- But independent of such a definition, I am terminally disorganised
- Seriously what the fuck is
../5-virus-mix/2016-10-11__ref896__reg2084-5083__sd1001 - I think command wrappers and platforms like
Galaxyget in the way of things too much - I wrote a “shell” to try and compensate for this
- Now I have a shell, it is called
chitin
-
This is a genuine directory in my file system, created about a month ago. It contains results for a run of
Gretelagainst thepolgene on theHIVgenome (2084-5083). Off the top of my head, I cannot recall whatsd100is, or whyregappears before the base positions. I honestly tried. ↩ ↩ - Because more things that are not my actual PhD is just what my PhD needs. ↩
- If it helps you, imagine some soft jazz playing to the sound of rain while I talk about this gruffly in the dark with a cigarette poking out of my mouth. Oh, and everything is in black and white. It’s bioinformatique noir. ↩
-
I’m quite pleased with this one, because I pretty much always forget to
timehow long my assemblies and alignments take. ↩