In a change to my usual essay length posts, I wanted to share a quick bowtie2 tip for relaxing the parameters of alignment. It’s no big secret that bowtie2 has these options, and there’s some pretty good guidance in the manual, too. However, we’ve had significant trouble in our lab finding a suitable set of permissive alignment parameters.
In the course of my PhD work on haplotyping regions of metagenomes, I have found that even using bowtie2‘s somewhat permissive --very-sensitive-local, that sequences with an identity to the reference of less than 90% are significantly less likely to align back to that reference. This is problematic in my line of work, where I wish to recover all of the individual variants of a gene, as the basis of my approach relies on a set of short reads (50-250bp) aligned to a position on a metagenomic assembly (that I term the pseudo-reference). It’s important to note that I am not interested in the assembly of individual genomes from metagenomic reads, but the genes themselves.
Recently, the opportunity arose to provide some evidence to this. I have some datasets which constitute “synthetic metahaplomes” that consist of a handful of arbitrary known genes that all perform the same function, each from a different organism. These genes can be broken up into synthetic reads and aligned to some common reference (another gene in the same family).
This alignment can be used a means to test my metagenomic haplotyper; Gretel (and her novel brother data structure, Hansel), by attempting to recover the original input sequences, from these synthetic reads. I’ve already reported in my pre-print that our method is at the mercy of the preceding alignment, and used this as the hypothesis for a poor recovery in one of our data sets.
Indeed as part of my latest experiments, I have generated some coverage heat maps, showing the average coverage of each haplotype (Y-axis) at each position of the pseudo-reference (X-axis) and I’ve found that for sequences beyond the vicinity of 90% sequence identity, --very-sensitive-local becomes unsuitable.
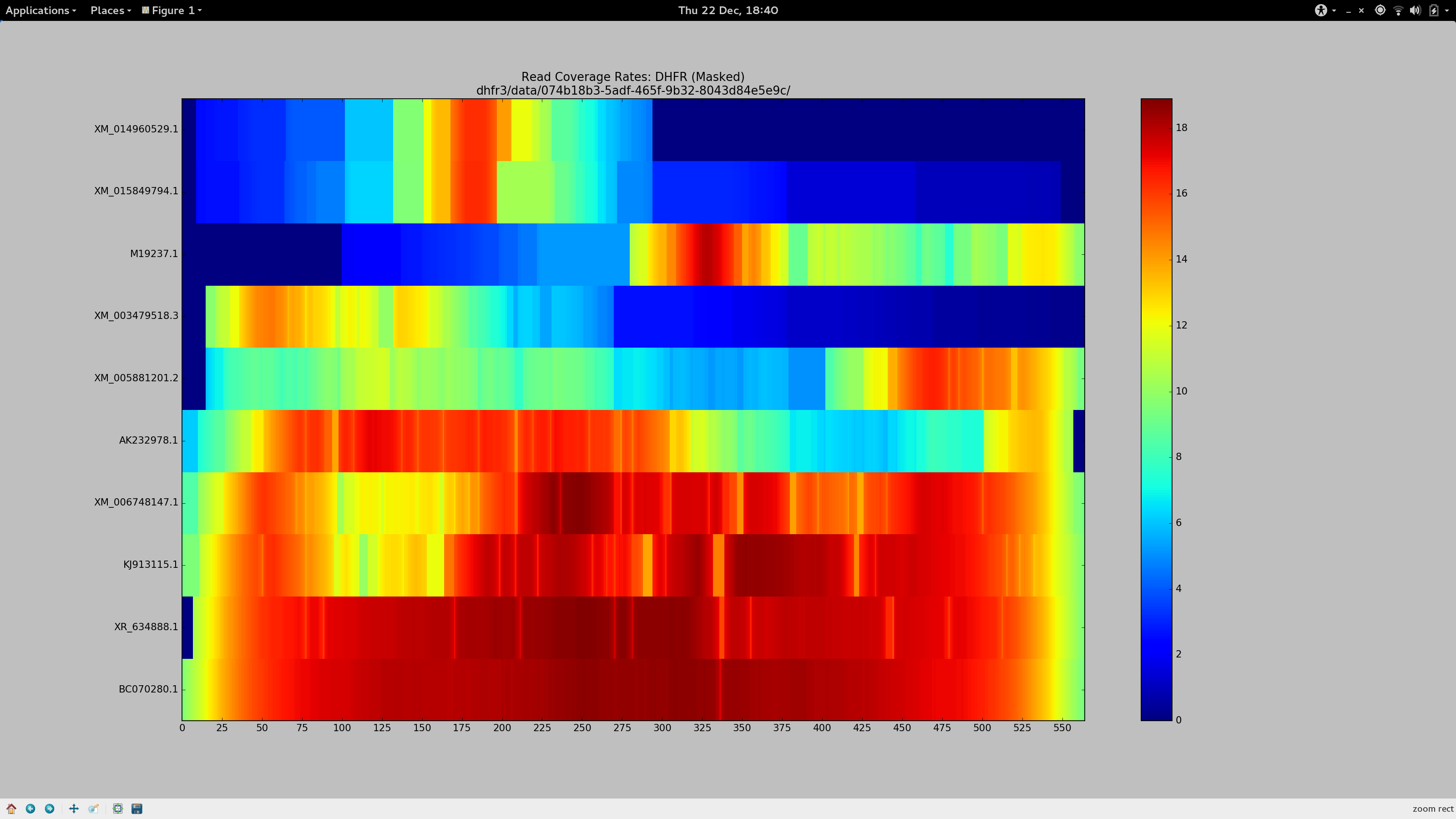
The BLAST record below represents the alignment that corresponds to the gene whose reads go on to align at the average coverage depicted at the top bar of the above heatmap. Despite its 79% identity, it looks good(TM) to me, and I need sequence of this level of diversity to align to my pseudo-reference so it can be included in Gretel‘s analysis. I need generous alignment parameters to permit even quite diverse reads (but hopefully not too diverse such that it is no longer a gene of the same family) to map back to my reference. Otherwise Gretel will simply miss these haplotypes.

So despite having already spent many days of my PhD repeatedly failing to increase my overall alignment rates for my metagenomes, I felt this time it would be different. I had a method (my heatmap) to see how my alignment parameters affected the alignment rates of reads on a per-haplotype basis. It’s also taken until now for me to quantify just what sort of sequences we are missing out on, courtesy of dropped reads.
I was determined to get this right.
Playing fucking mastermind with bowtie2 parameters pic.twitter.com/yDHduJPBNM
— Sam Nicholls (@samstudio8) December 23, 2016
For a change, I’ll save you the anticipation and tell you what I settled on after about 36 hours of getting cross.
--local -D 20 -R 3
Ensure we’re not performing end-to-end alignment (allow for soft clipping and the like), and borrow the most sensitive default “effort” parameters.-L 3
The seed substring length. Decreasing this from the default (20 - 25) to just3allows for a much more aggressive alignment, but adds computational cost. I actually had reasonably good results with-L 11, which might suit you if you have a much larger data set but still need to relax the aligner.-N 1
Permit a mismatch in the seed, because why not?--gbar 1
Has a small, but noticeable effect. Appears to thin the width of some of the coverage gap in the heatmap at the most stubborn sites.--mp 4
Reduces the maximum penalty that can be applied to a strongly supported (high quality) mismatch by a third (from the default value of6). The aggregate sum of these penalties are responsible for the dropping of reads. Along with the substring length, this had a significant influence on increasing my alignment rates. If your coverage stains are stubborn, you could decrease this again.
Tada.
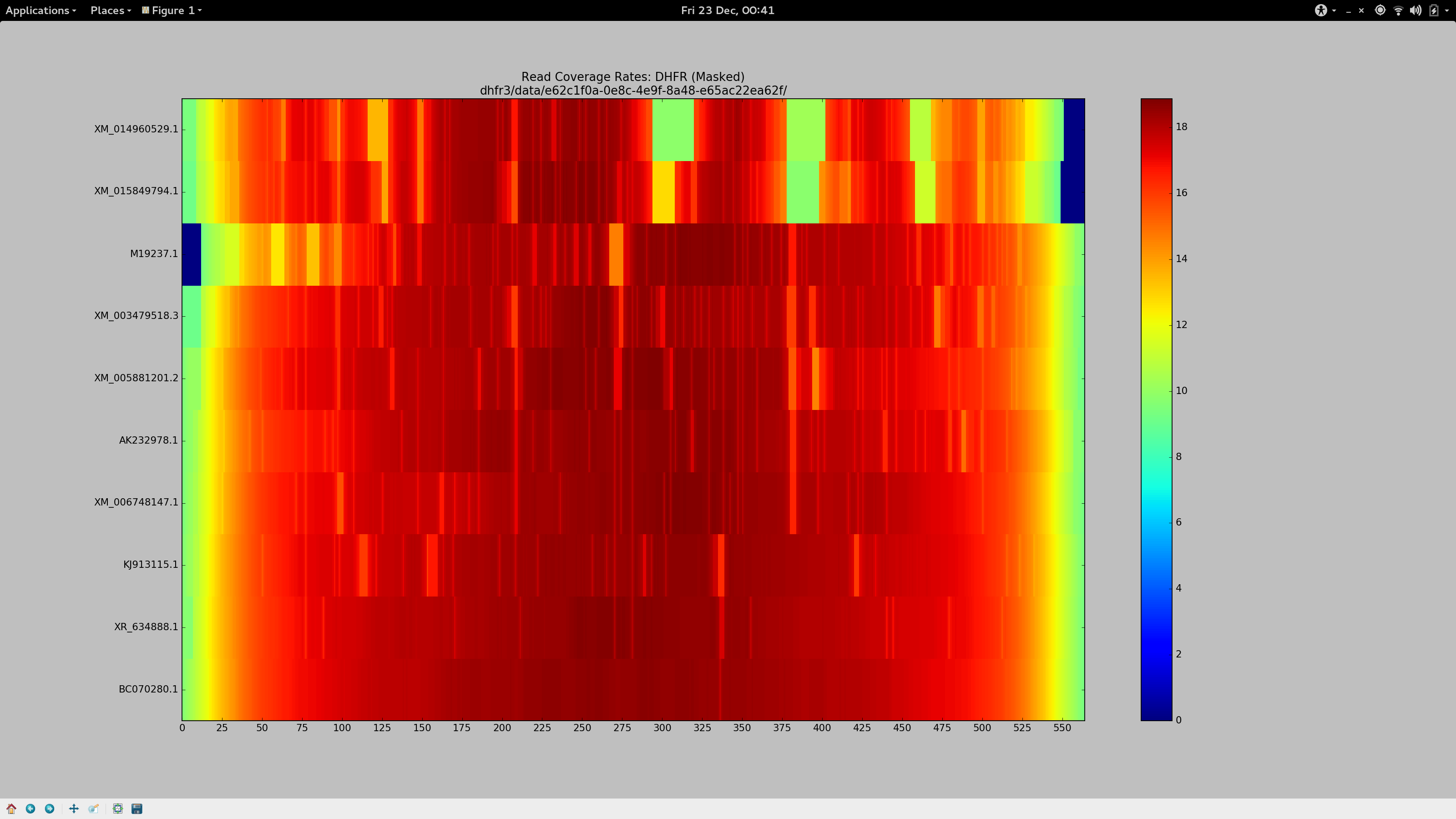
tl;dr
bowtie2 --local -D 20 -R 3 -L 3 -N 1 -p 8 --gbar 1 --mp 3