In the opening act of this status report, I described the abrupt and unfortunate end to the adventure of my pre-print. In response to reviewer feedback, I outlined three major tasks that lie ahead in wait for me, blocking the path to an enjoyable Christmas holiday, and a better manuscript submission to a more specialised an alternative journal:
- Improve Triviomes
We are already doing something interesting and novel, but the “triviomes” are evidently convoluting the explanation. We need something with more biological grounding such that we don’t need to spend many paragraphs explaining why we’ve made certain simplifications, or cause readers to question why we are doing things in a particular way. Note this new method will still need to give us a controlled environment to test the limitations of
Hansel and Gretel.
- Polish DHFR and AIMP1 analysis
One of our reviewers misinterpreted some of the results, and drew a negative conclusion aboutGretel‘s overall accuracy. I’d like to revisit the DHFR and AIMP1 data sets to both improve the story we tell, but also to describe in more detail (with more experiments) under what conditions we can and cannot recover haplotypes accurately.
- Real Reads
Create and analyse a data set consisting of real reads.
The first part of this report covers my vented frustrations with both generating, but particularly analysing the new “Treeviomes” in depth. This part will now turn focus to the sequel of my already existing DHFR analysis, addressing the reasoning behind a particularly crap set of recoveries and reassuring myself that perhaps everything is not a disaster after all.
DHFR II: Electric Boogaloo
Flashback
Our preprint builds upon the initial naive testing of Hansel and Gretel on recovery of randomly generated haplotypes, by introducing a pair of metahaplomes that each contain five haplotypes of varying similarity of a particular gene; namely DHFR and AIMP1.

Testing follows the same formula as we’ve discussed in a previous blog post:
- Select a master gene (an arbitrary,
DHFR, in this case) megaBLASTfor similar sequences and select5arbitrary sequences of decreasing identity- Extract the overlapping regions on those sequences, these are the input haplotypes
- Generate a random set of (now properly) uniformly distributed reads, of a fixed length and coverage, from each of the input haplotypes
- Use the master as a pseudo-reference against which to align your synthetic reads
- Stand idly by as many of your reads are hopelessly discarded by your favourite aligner
- Call for variants on what is left of your aligned reads by assuming any heterogeneous site is a variant
- Feed the aligned reads and called variant positions to
Gretel - Compare the recovered haplotypes, to the input haplotypes, with
BLAST

As described in our paper, results for both the DHFR and AIMP1 data sets were promising, with up to 99% of SNPs correctly recovered to successfully reconstruct the input haplotypes. We have shown that haplotypes can be recovered from short reads that originate from a metagenomic data set. However, I want to revisit this analysis to improve the story and fix a few outstanding issues:
- A reviewer assumed lower recovery rates on haplotypes with poorer identity to the pseudo-reference was purely down to bad decision making by
Gretel, and not to do with the long paragraph about how this is caused by discarded reads - A reviewer described the tabular results as hard to follow
- I felt I had not provided a deep enough analysis to readers into the effects of read length and coverage, although no reviewer asked for this, it will help me sleep at night
- Along the same theme, the tables in the pre-print only provided averages, whereas I felt a diagram might better explain some of the variance observed in recovery rates
- Additionally, I’d updated the read generator as part of my work on the “Treeviomes” discussed in my last post, particularly to generate more realistic looking distributions of reads, I needed to check results were still reliable and thought it would be helpful if we had a consistent story between our data sets
Progress
Continuing the rampant pessimism that can be found in the first half of this status report, I wanted to audit my work on these “synthetic metahaplomes”, particularly given the changed results between the Triviomes, and more biologically realistic and tangible Treeviomes. I wanted to be sure that we had not departed from the good results introduced in the DHFR and AIMP1 sections of the paper.
To be a little less gloomy, I was also inspired by the progress I had made on the analysis of the Treeviomes. I felt we could we could try and uniformly present our results and use box diagrams similar to those that I had finally managed to design and plot at the end of my last post. I feel these diagrams provide a much more detailed insight to the capabilities of Gretel given attributes of read sets such as length and coverage. Additionally, we know each of the input haplotypes, which unlike the uniformly varying sequences of our Treeviomes, have decreasing similarity to the pseudo-reference; thus we can use such a plot to describe Gretel‘s accuracy with respect to each haplotype’s distance from the pseudo-reference (somewhat akin to how we present results for different values of per-haplotype per-base sequence mutation rates on our Treeviomes).
So here’s one I made earlier:
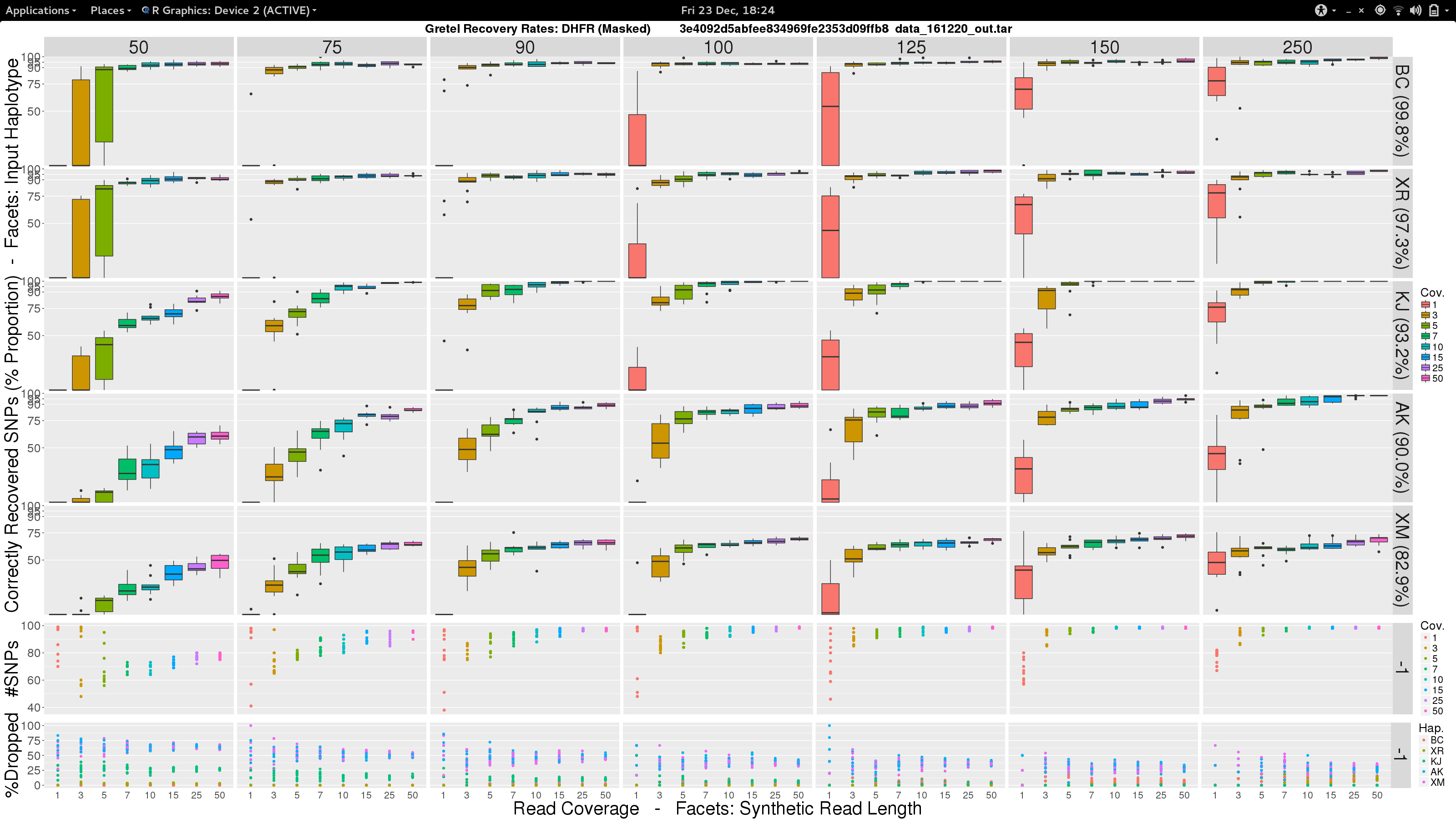
Sam, what the fuck am I looking at?
- Vertical facets are read length
- Horizontal facets represent each of the five input haplotypes, ordered by decreasing similarity to the chosen
DHFRpseudo-reference - X-axis of each boxplot is the average read coverage for each of the ten generated read sets
- Y-axis is the average best recovery rate for a given haplotype, over the ten runs of
Gretel: each boxplot summarising the best recoveries for ten randomly generated uniform distributed sets of reads (with a set read length and coverage) - Top scatter plot shows number of called variants across the ten sets of reads for each length-coverage parameter pair
- Bottom scatter plot shows for each of the ten read sets, the proportion of reads from each haplotype that were dropped during alignment
What can we see?
- Everything is not awful: three of the five haplotypes can be recovered to around 95% accuracy, even with very short reads (75-100bp), and reasonable average per-haplotype coverage.
- Good recovery of
AK232978, despite a 10% mutation rate when compared to the pseudo-reference:Gretelyielded haplotypes with 80% accuracy. - Recovery of
XM_012113510is particularly difficult. Even given long reads (150-250bp) and high coverage (50x) we fail to reach 75% accuracy. Our pre-print hypothesiszed that this was due to its 82.9% identity to the pseudo-reference causing dropped reads.
Our pre-print presents results considering just one read set for each of the length and coverage parameter pairs. Despite the need to perhaps take those results with a pinch of salt, it is somewhat relieving to see that our new more robust experiment yields very similar recovery rates across each of the haplotypes. With the exception of XM_012113510.
The Fall of “XM”
Recovery rates for our synthetic metahaplomes, such as DHFR, are found by BLASTing the haplotypes returned by Gretel against the five known, input haplotypes. The “best haplotype” for an input is defined as the output haplotype with the fewest mismatches against the given input. We report the recovery rate by assuming the mismatches are the incorrectly recovered variants, and divide this by the number of variants called over the read set that yielded this haplotype.
We cannot simply use sequence identity as a score: consider a sequence of 100bp, if there are just 5 variants, all of which are incorrectly reconstructed, Gretel will still appear to be 95% accurate, rather than correctly reporting 0% (1 – 5/5). Note however, that our method does somewhat assume that variants are called perfectly: remember that homogenous sites are ignored by Gretel, so any “missed” SNPs, will always mismatch the input haplotypes, as we “fill in” homogeneous sites when outputting the output haplotype FASTA, with the nucleotides from the pseudo-reference.
XM_012113510 posed an interesting problem for evaluation. When collating results for the pre-print, I found the corresponding best haplotype failed to align the entirity of the gene (564bp) and instead yielded two high scoring, non-overlapping hits. When I updated the harness that calculates and collates recovery rates across all the different runs of Gretel, I overlooked the possibility for this and forced selection of the “best” hit (-max_hsps 1). Apparently, this split-hit behaviour was not a one-off, but almost universal across all runs of Gretel over the 640 sets of reads.
In fact, the split-hit was highly precise. Almost every best XM_012113510 haplotype had a hit along the first 87bp, and another starting at (or very close to) 135bp, stretching across the remainder of the gene. My curiosity piqued, I fell down another rabbit hole.
Curious Coverage
I immediately assumed this must be an interesting side-effect of read dropping. I indicated in our pre-print that we had difficulty aligning reads against a pseudo-reference where those reads originate from a sequence that is dissimilar (<= 90% identity) from the reference. This read dropping phenomenon has been one of the many issues our lab has experiened with current bioinformatics tooling when dealing with metagenomic data sets.
Indeed, our boxplot above features a colour-coded scatter plot that demonstrates that the two most dissimilar input haplotypes AK and XM, are also the haplotypes that experience the most trouble during read alignment. I suggested in the pre-print that thes dropped reads are the underlying reason for Gretel‘s performance on those more dissimilar haplotypes.
I wanted to see empirically whether this was the case, and to potentially find a nice way of providing evidence, given that one of our reviewers missed my attribution of these poorer AK and XM recoveries to trouble with our alignment step, rather than incompetence on Gretel‘s part. I knocked up a small Python script that deciphered my automatically generated read names, and calculated per-haplotype read coverage, for each of the five input haplotypes, across the 640 alignments.
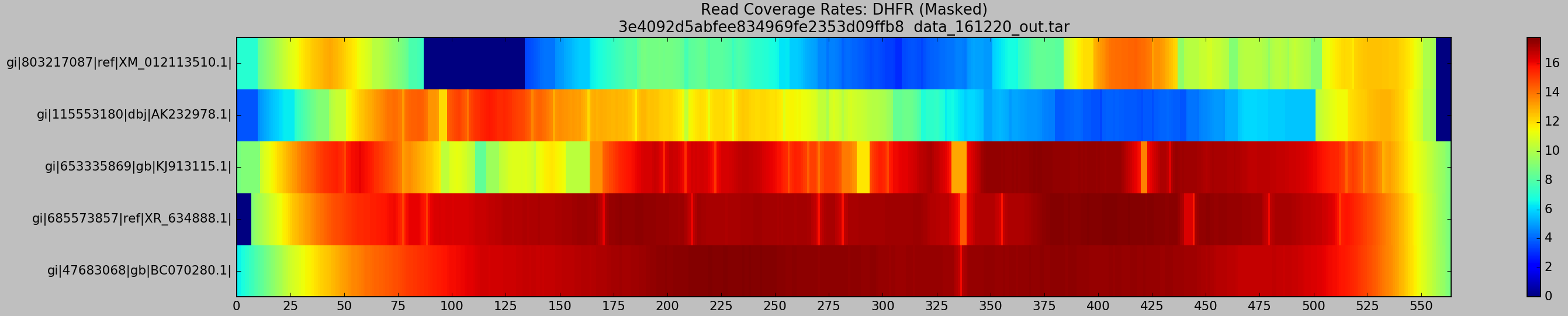
- X-axis represents genomic position along the psuedo-reference (the “master” `DHFR gene)
- Y-axis labels each of the input haplotype, starting with the most dissimilar (
XM) and ending on the most similar (BC) - The coloured heatmap plots the average coverage for a given haplotype at a given position, across all generated BAM files
Well look what we have here… Patches of XM and AK are most definitely poorly supported by reads! This is troubling for their recovery, as Hansel stores pairwise evidence of variants co-occurring on the same read. If those reads are discarded, we have no evidence of that haplotype. It should be no surprise that Gretel has difficultly here. We can’t create evidence from nothing.
What’s this?… A crisp, ungradiated, dark navy box representing absolutely 0 reads across any of the 640 alignments, sitting on our XM gene. If I had to guesstimate the bounds of that box on the X-axis, I would bet my stipend that they’ll be a stonesthrow from 87 and 135bp… The bounds of the split BLAST hits we were reliably generating against XM_012113510, almost 640 times. I delved deeper. What did the XM reads over that region look like? Why didn’t they align correctly? Where they misaligned, or dropped entirely?
But I couldn’t find any.
Having a blast with BLAST
After much head scratching. I began an audit from the beginning, consulting the BLAST record that led me to select XM_012113510.1 for our study:
|
1 |
EU145592.1 XM_012113510.1 82.979 564 46 1 1 564 52 565 3.89e-175 625 |
Seems legit? An approximately 83% hit with full query coverage (564bp) consisting of 46 mismatches and containing an indel of length one? Not quite, as I would later find. But to cut an already long story somewhat shorter. The problem boils down to me being unable to read, again.
BLASToutfmt6 alignment lengths include deletions on the subjectBLASToutfmt6 tells you the number of gaps, not their size
For fuck’s sake, Sam. Sure enough, here’s the full alignment record. Complete with a fucking gigantic 50bp deletion on the subject, XM_012113510.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 |
Query 1 ATGGTTGGTTCGCTAAACTGCATCGTCGCTGTGTCCCAGAACATGGGCATCGGCAAGAAC 60 |||||| || |||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct 52 ATGGTTCGTCCGCTAAACTGCATCGTCGCTGTGTCCCAGAACATGGGCATCGGCAAGAAC 111 Query 61 GGGGACCTGCCCTGGCCACCGCTCAGGAATGAATTCAGATATTTCCAGAGAATGACCACA 120 ||| |||||||||||||||| ||||| Sbjct 112 GGGAACCTGCCCTGGCCACCACTCAG---------------------------------- 137 Query 121 ACCTCTTCAGTAGAAGGTAAACAGAATCTGGTGATTATGGGTAAGAAGACCTGGTTCTCC 180 ||||||||||| ||||||||||||||| |||||||||||||||| Sbjct 138 ----------------GTAAACAGAATTTGGTGATTATGGGTAGGAAGACCTGGTTCTCC 181 Query 181 ATTCCTGAGAAGAATCGACCTTTAAAGGGTAGAATTAATTTAGTTCTCAGCAGAGAACTC 240 ||||| |||||||||||||||||||||| ||||||||| |||||||||| ||||||||| Sbjct 182 ATTCCAGAGAAGAATCGACCTTTAAAGGACAGAATTAATATAGTTCTCAGTAGAGAACTC 241 Query 241 AAGGAACCTCCACAAGGAGCTCATTTTCTTTCCAGAAGTCTAGATGATGCCTTAAAACTT 300 |||||||||||| | ||||||||||||||| ||| |||||| |||||||||||| ||||| Sbjct 242 AAGGAACCTCCAAAGGGAGCTCATTTTCTTGCCAAAAGTCTGGATGATGCCTTAGAACTT 301 Query 301 ACTGAACAACCAGAATTAGCAAATAAAGTAGACATGGTCTGGATAGTTGGTGGCAGTTCT 360 | |||| | ||||||||| ||||||||||||||||||| |||||||| || ||||||||| Sbjct 302 ATTGAAGATCCAGAATTAACAAATAAAGTAGACATGGTTTGGATAGTGGGAGGCAGTTCT 361 Query 361 GTTTATAAGGAAGCCATGAATCACCCAGGCCATCTTAAACTATTTGTGACAAGGATCATG 420 || ||||||||||||||||| | ||||||||||||| |||||||||||||||||||||| Sbjct 362 GTATATAAGGAAGCCATGAACAAGCCAGGCCATCTTAGACTATTTGTGACAAGGATCATG 421 Query 421 CAAGACTTTGAAAGTGACACGTTTTTTCCAGAAATTGATTTGGAGAAATATAAACTTCTG 480 ||||| ||||||||||| |||||| |||||||||||||| || |||||||||||||| Sbjct 422 CAAGAATTTGAAAGTGATGTGTTTTTCCCAGAAATTGATTTTGAAAAATATAAACTTCTT 481 Query 481 CCAGAATACCCAGGTGTTCTCTCTGATGTCCAGGAGGAGAAAGGCATTAAGTACAAATTT 540 |||||||| |||||||||| | |||||||||||||| ||||||||||||||||||||| Sbjct 482 CCAGAATATCCAGGTGTTCCTTTGGATGTCCAGGAGGAAAAAGGCATTAAGTACAAATTT 541 Query 541 GAAGTATATGAGAAGAATGATTAA 564 ||||||||||| ||||| ||||| Sbjct 542 GAAGTATATGAAAAGAACAATTAA 565 |
It is no wonder we couldn’t recover 100% of the XM_012113510 haplotype, when compared to our pseudo-reference, 10% of it doesn’t fucking exist. Yet, it is interesting to see that the best recovered XM_012113510‘s were identified by BLAST to be very good hits to the XM_012113510 that actually exists in nature, despite my spurious 50bp insertion. Although Gretel is still biased by the structure of the pseudo-reference, (which is one of the reasons that insertions and deletions are still a pain), we are still able to make accurate recoveries around straightforward indel sites like this.
As lovely as it is to have gotten to the bottom of the XM_012113510 recovery troubles, this doesn’t really help our manuscript. We want to avoid complicated discussions and workarounds that may confuse or bore the reader. We already had trouble with our reviewers misunderstanding the purpose of the convoluted Triviome method and its evaluation. I don’t want to have to explain why recovery rates for XM_012113510 need special consideration because of this large indel.
I decided the best option was to find a new member of the band.
DHFR: Reunion Tour
As is the case whenever I am certain something won’t take the entire day, this was not as easy as I anticipated. Whilst it was trivial to BLAST my chosen pseudo-reference once more and find a suitable replacement (reading beyond “query cover” alone this time), trouble arose once more in the form of reads dropped via alignment.
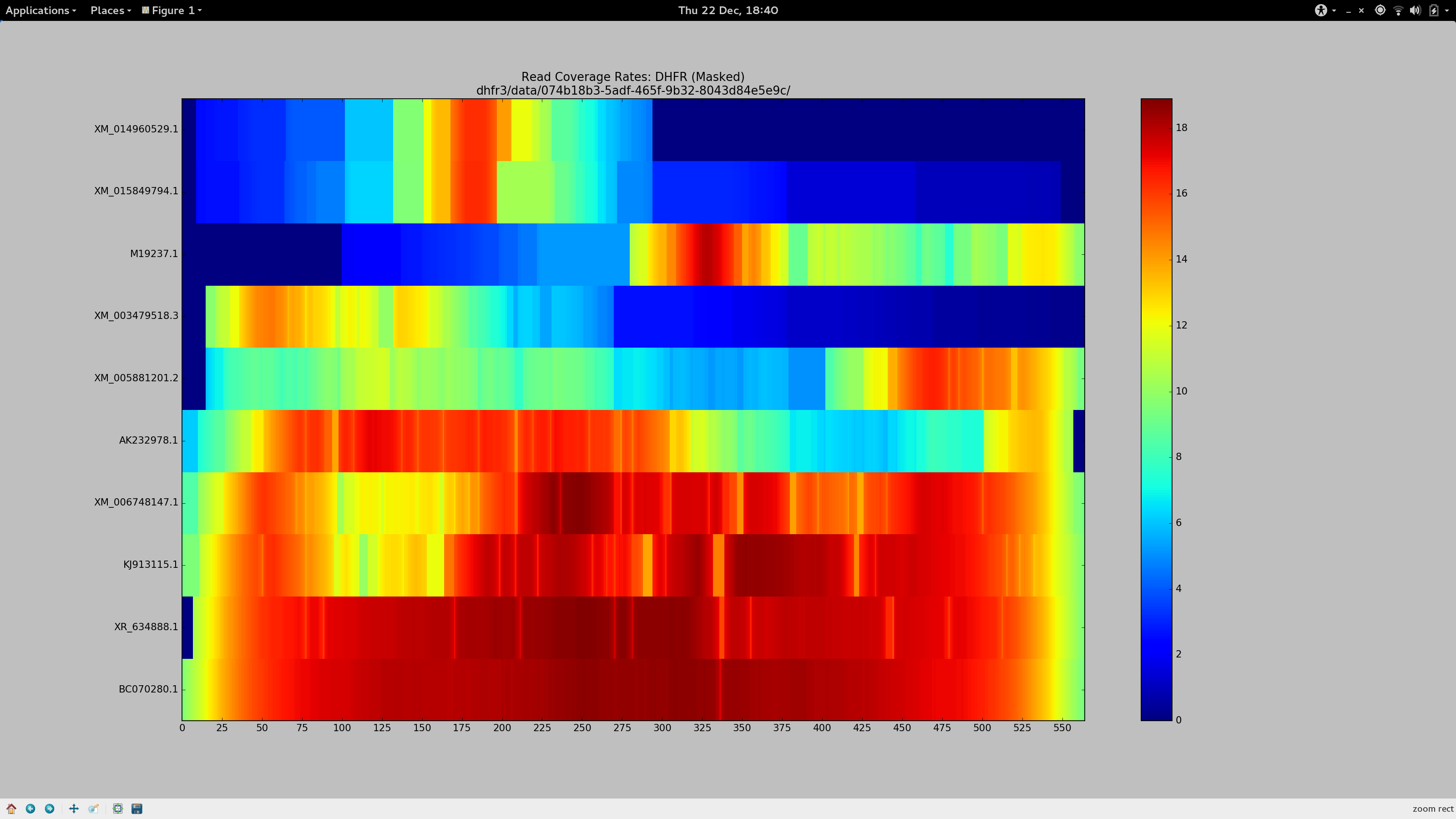
Determined that these sequences should (and must) align, I dedicated the day to finally find some bowtie2 parameters that would more permissively align sequences to my pseudo-reference to ensure Gretel had the necessary evidence to recover even the more dissimilar sequences:
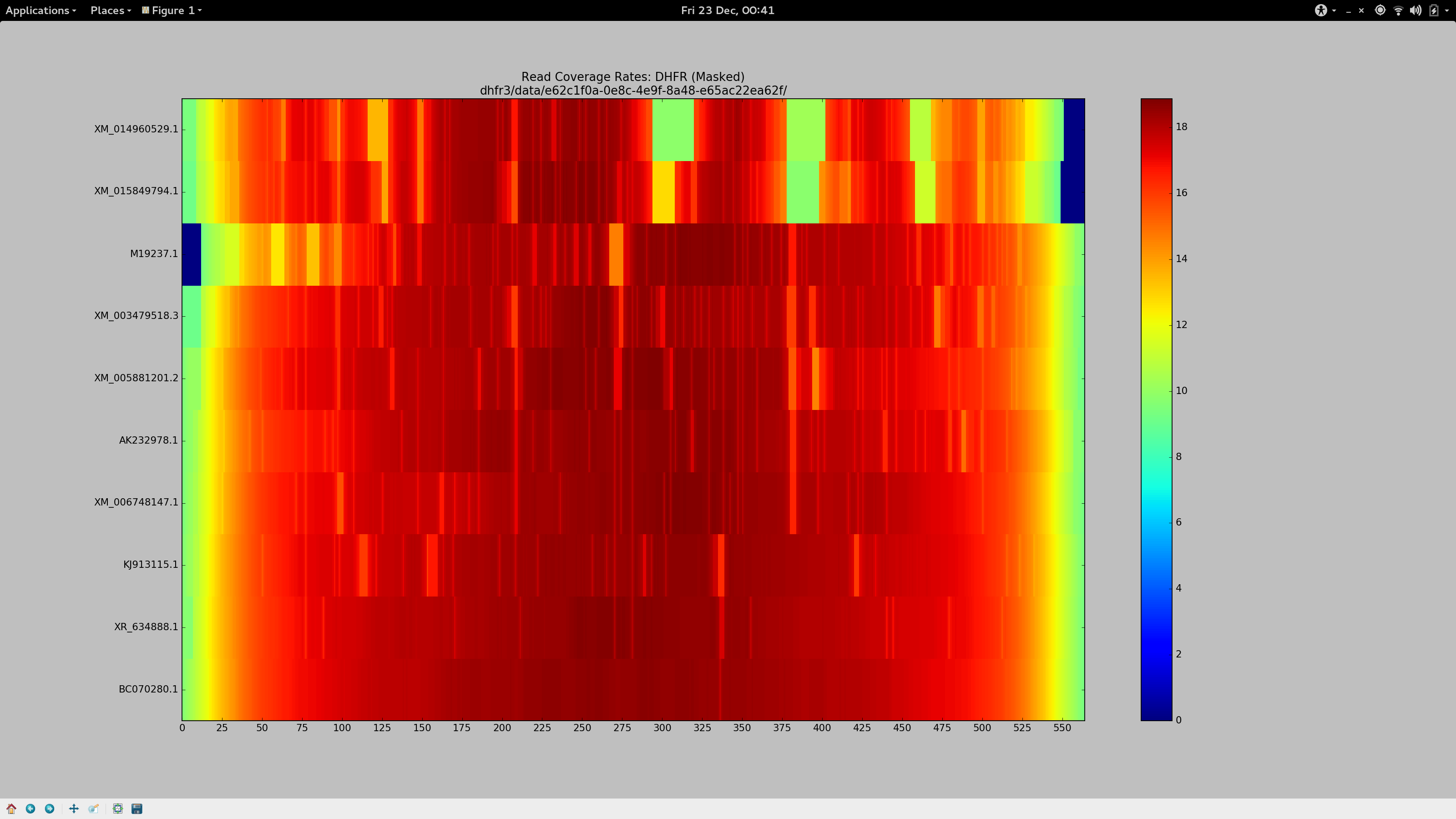
Success.
So, what’s next?
Wonderful. So after your productive day of heatmap generation, what are you going to do with your life new found alignment powers?
✓ Select a new DHFR line-up
The door has been blown wide open here. Previously our input test haplotypes had a lower bound of around 90% sequence similarity to avoid the loss of too many reads. However with my definition that I have termed --super-sensitive-local, we can attempt recoveries of genes that are even more dissimilar from the reference! I’ve actually already made the selection, electing to keep BC (99.8%), XR (97.3%) and AK (90.2%). I’ve remove KJ (93.2%) and the troublemaking XM (85.1%) to make way for the excitingly dissimilar M (83.5%) and (a different) XM (78.7%).
✓ Make a pretty picture
After some chitin and Sun Grid Engine wrangling, I present this:
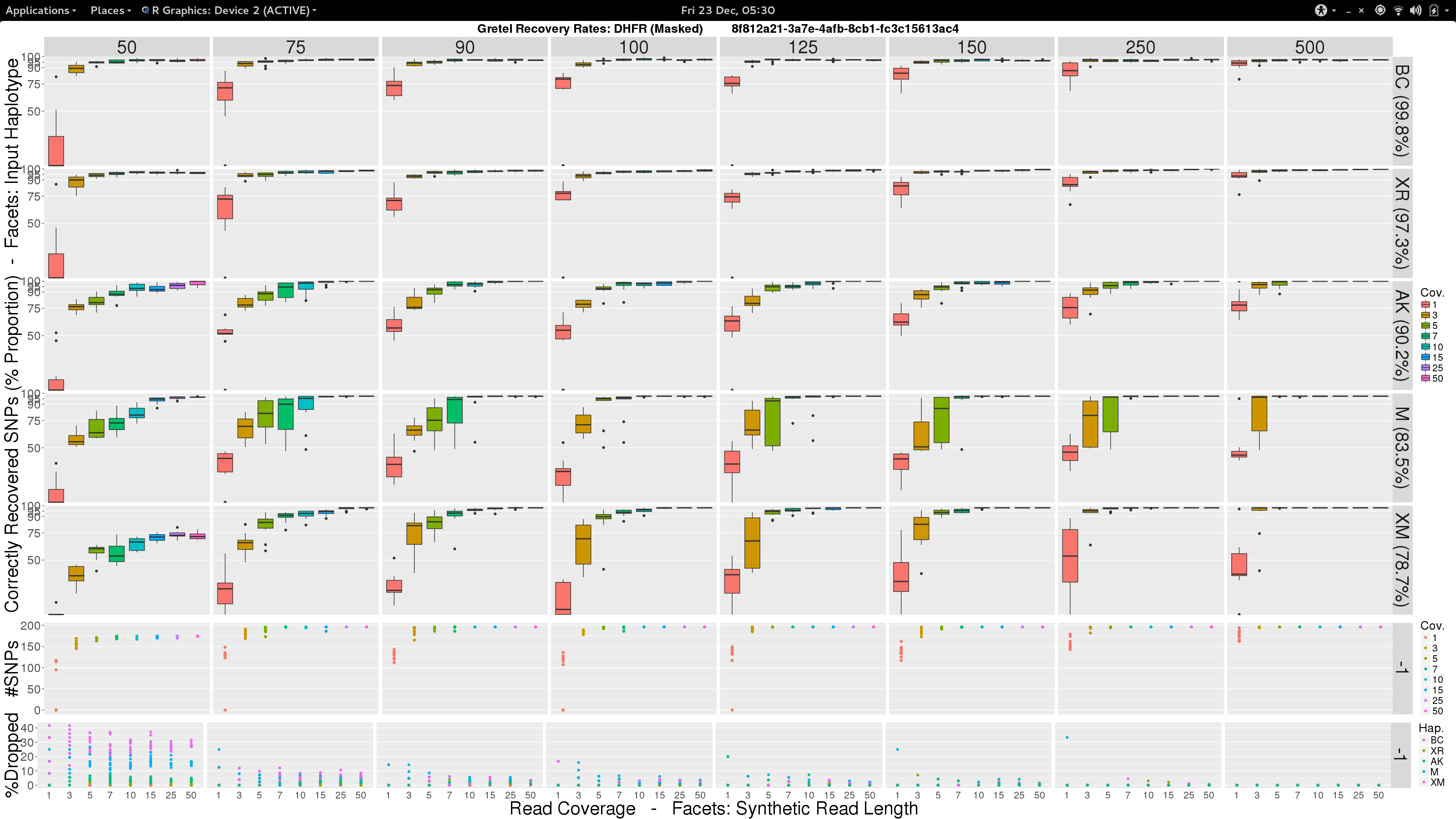
✓ Stare in disbelief
Holy crap. It works.
Conclusion
- Find what is inevitably wrong with these surprisingly excellent results
- Get this fucking paper done
- Merry Christmas
tl;dr
- In a panic I went back to check my
DHFRanalysis and then despite it being OK, changed everything anyway - One of the genes from our paper is quite hard to recover, because 10% of it is fucking missing
- Indels continue to be a pain in my arse
- Contrary to what I said last week, not everything is awful
- Coverage really is integral to
Gretel‘s ability to make good recoveries - I’ve constructed some parameters that allow us to permissively align metagenomic reads to our pseudo-reference and shown it to massively improve
Gretel‘s accuracy bowtie2needs some love <3- I probably care far too much about integrity of data and results and should just write my paper already
- A lot of my problems boil down to not being able to read
- I owe Hadley Wickham a drink to making it so easy to make my graphs look so pretty
- Everyone agrees this should be my last Christmas as a PhD
- Tom is mad because I keep telling him about bugs and pitfalls I have find in bioinformatics tools